

DImEnsION @ Warwick: a research group led by Dr. G.C. Sosso, focussing on Molecular Simulations of Disordered Systems and Phase Transitions.
We use Computational Methods to investigate the functional properties of systems of practical relevance, such as water and ice or amorphous solids of interest for electronic devices.

I am a computational scientist, chiefly interested in the physical chemistry of condensed matter, from supercooled liquids to biological interfaces. My vision is to understand the chemistry, the functional properties and the phase transitions of different classes of systems, from metallic glasses to cellular membranes. My approach consists in performing fundamental research using computer simulations, aimed at rationalise, complement and guide experiments and applications.
DImEnsION @ Warwick focuses onMolecular Simulations of Disordered Systems and Phase Transitions.

News
People

Publications

Research

Teaching
Outreach
Join
Case study – The Formation of Ice in Biological Matter
One of the main research interests of the SossoGroup is the formation of ice in biological matter. In 2022, the group as been awarded, as part of collaborative effort involving a number of experimental partners, the Horizon Prize from the Royal Society of Chemistry, for “the development, application and translation of chemical tools for cryobiology”. The animation below contains a brief summary of why understanding and controlling ice formation is key in the context of cryopreservation – and what contributions we have made in that regard.
Case study
The formation of ice on cholesterol crystals: unravelling the molecular-level details by means of a combined effort of experiments and simulations. Check out the whole story – published in an open access article available here. Thanks to the Royal Society of Chemistry for condensing our work into a snazzy video!
Stay in touch with us via Twitter @SossoGroup
Super excited that we will be hosting the @SLTBCryo meeting here @UoMMIB in September, with #team_ice @SossoGroup @TF_Whale ! For all #cryobiology and low temperature bioscience it will be a MUST attend event.
Fabulous news for @warwickchem, @WarwickCaTCh and Chemistry co-lead @lbpartay that the @EPSRC funded @HetSysCDT will be training a new generation of scientists in computational modelling to tackle pressing global sustainability challenges! #compchem
Come and join us to develop molecular models for next generation battery interfaces!! We have a fully funded 4 yr @EPSRC PhD studentship open to UK and overseas candidates. Students will receive training and will be based @MIF_UoL @livunieng @livunisire
Very excited to be able to share the news that the @HetSysCDT has been renewed for a further 5 years! HetSys II will be led by me in @WarwickEngineer, @nickhine82 in @WarwickPhysics and @lbpartay in @warwickchem plus a team from 4 further depts and 46 external partners
Recent Publications
2023
Understanding the impact of ammonium ion substitions on heterogeneous ice nucleation Journal Article Forthcoming
In: Faraday Discussions, Forthcoming.
Combining machine learning and molecular simulations to predict the stability of amorphous drugs Journal Article
In: The Journal of Chemical Physics, vol. 159, no. 1, pp. 014503, 2023.
Interplay of multiple clusters and initial interface positioning for forward flux sampling simulations of crystal nucleation Journal Article
In: The Journal of Chemical Physics, vol. 158, no. 22, pp. 224102, 2023.
Generating protein folding trajectories using contact-map-driven directed walks Journal Article
In: Journal of Chemical Information and Modeling, vol. 63, no. 7, pp. 2181–2195, 2023.
Understanding the emergence of the boson peak in molecular glasses Journal Article
In: Nature Communications, vol. 14, no. 1, pp. 1–11, 2023.
2022
Leveraging genetic algorithms to maximise the predictive capabilities of the SOAP descriptor Journal Article
In: Molecular Systems Design & Engineering, 2022.
Minimalistic ice recrystallisation inhibitors based on phenylalanine Journal Article
In: Chemical Communications, vol. 58, no. 55, pp. 7658–7661, 2022.
The role of structural order in heterogeneous ice nucleation Journal Article
In: Chemical Science, vol. 13, iss. 17, pp. 5014-5026, 2022.
Ice recrystallization inhibition by amino acids: the curious case of alpha- and beta-alanine Journal Article
In: The Journal of Physical Chemistry Letters, vol. 13, no. 9, pp. 2237-2244, 2022.
Lipid bilayers as potential ice nucleating agents Journal Article
In: Physical Chemistry Chemical Physics, vol. 24, iss. 11, pp. 6476-6491, 2022.
How do interfaces alter the dynamics of supercooled water? Journal Article
In: Nanoscale, vol. 14, iss. 11, pp. 4254-4262, 2022.
2021
Recovering local structure information from high-pressure total scattering experiments Journal Article
In: Journal of Applied Crystallography, vol. 54, no. 6, pp. 1546-1554, 2021.
The seven deadly sins: When computing crystal nucleation rates, the devil is in the details Journal Article
In: The Journal of Chemical Physics, vol. 155, no. 4, pp. 040901, 2021.
A minimalistic cyclic ice-binding peptide from phage display Journal Article
In: Nature Communications, vol. 12, pp. 2675, 2021.
Microscopic Kinetics Pathway of Salt Crystallization in Graphene Nanocapillaries Journal Article
In: Physical Review Letters, vol. 126, no. 13, pp. 136001, 2021.
Modelling the interactions and diffusion of NO in amorphous SiO2 Journal Article
In: Modelling and Simulation in Materials Science and Engineering, vol. 29, no. 3, pp. 035008, 2021.
The atomistic details of the ice recrystallisation inhibition activity of PVA. Journal Article
In: Nature Communications, vol. 12, no. 1323, 2021.
2020
Combining high-resolution scanning tunnelling microscopy and first-principles simulations to identify halogen bonding Journal Article
In: Nature Communications, vol. 11, pp. 2103, 2020.
Insights into the Emerging Networks of Voids in Simulated Supercooled Water Journal Article
In: The Journal of Physical Chemistry B, vol. 124, no. 11, pp. 2180-2190, 2020.
Less may be more: an informed reflection on molecular descriptors for drug design and discovery Journal Article
In: Molecular Systems Design and Engineering, vol. 5, pp. 317-329, 2020.
Atomistic simulations of thermal conductivity in GeTe nanowires Journal Article
In: Journal of Physics D: Applied Physics, vol. 53, pp. 054001, 2020.

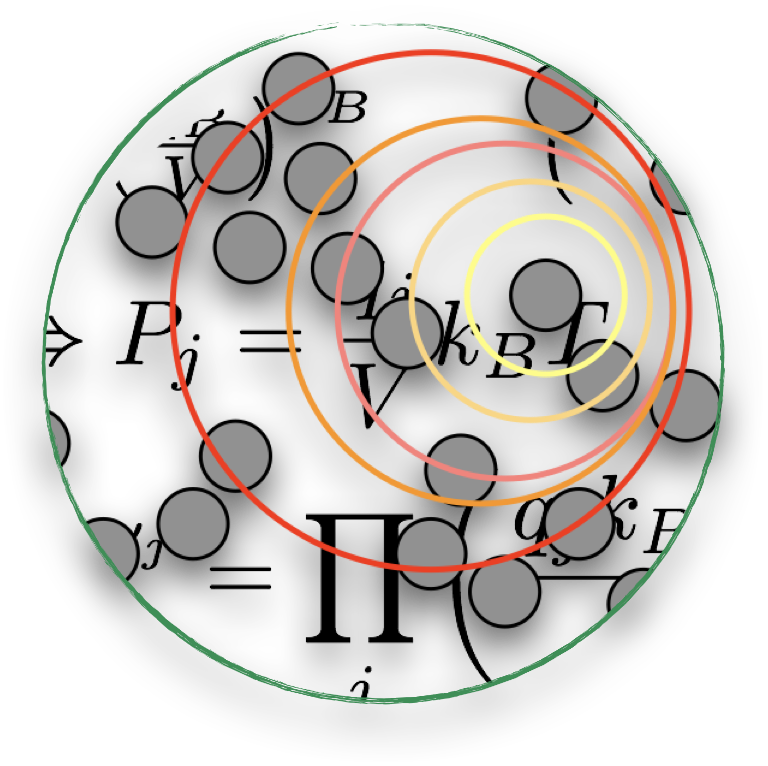
Gabriele teaches Statistical Mechanics – that branch of physics connecting the microscopic world (stuff best dealt with via Quantum Mechanics) and the macroscopic world (stuff described by Thermodynamics).
Incidentally, Statistical Mechanics also represents the theoretical foundations of much of the computational work taking place at DImEnsION @ Warwick…
On top of that, Gabriele will soon lecture about Thermodynamics as well, and he will also deliver part of a module ominously named “Advanced Computational Chemistry”.
Everything relevant to the Statistical Mechanics module can be found on Moodle Warwick, but check out our Teaching page for selected material…
Most importantly, do remember…

A number of opportunities to join DImEnsION @ Warwick are presently availabe:

Want to know more? Thinking about getting involved? Drop us a line at [email protected].