µCell - Interdisciplinary Research in Modelling and Simulation of Cell Spatial Behaviour
by Dominic Orchard, Jonathan Gover, Lee Lewis Herrington, James Lohr, Duncan Stead, Cathy Young and Sara Kalvala,[1] Department of Computer Science, University of Warwick
Abstract
A central aim of systems biology is the strengthening of quantitative and qualitative knowledge of biological systems by studying the interactions between components and processes that lead to emerging properties and behaviours. Systems biology proliferated over the latter half of the twentieth century with the aid of technological advances and subsequent interdisciplinary research between natural scientists, computer scientists, and mathematicians. In this paper we present μCell, an interdisciplinary research project undertaken by undergraduates at the University of Warwick, seeking to aid systems biology intuition. The project's main contribution is a modelling and simulation tool for multi-cellular environments aimed at simulating higher-level cellular behaviours based on the interoperation of biochemical signalling pathway models and procedural models of cell components and structures, such as flagella. Based on these interoperated models, μCell is able to simulate spatial properties and behaviours of cells, such as chemotaxis. This paper introduces μCell, gives a case study model and simulation of flagella-based chemotaxis in E. coli, and discusses the pedagogical outcomes of the project for the students.
Keywords: μCell, systems biology, biological modelling, simulation, model interoperation, chemotaxis.
Introduction
Technological and scientific progress has yielded successively higher resolution techniques for the observation and manipulation of cells, aiding biological research. However, there is still much to be understood. The interaction of cells with their environment and with each other, via processes such as adhesion, movement, and quorum sensing, induces behaviour such as cell migration, aggregation of cells into tissues, precision growth across relatively large distances, and group behaviour such as fruiting body formation (Hynes and Lander, 1992). A thorough understanding of the cause and control of such behaviours is difficult due to the complexity of cells and their interactions.
Studying the components and processes of a cell independently from other components and processes often fails to expose the full spectrum of a cell's properties and behaviour. The study of emergent behaviours and properties at the cellular and organismal level requires an understanding of the dynamic interactions and causal relationships between individual lower-level processes within a cell, and between cells. Such study is the focus of the burgeoning field of systems biology (Kitano, 2002). In the last decade, interdisciplinary research between natural scientists, computer scientists, and mathematicians has greatly improved understanding through computational analyses, modelling, and simulation.
We present here the μCell modelling and simulation tool for multi-cellular environments where spatial behaviours can be simulated from models of biochemical signalling pathways interoperated with abstract procedural models of cell components, such as flagella. The models are procedural in the sense that they describe the operations of a component in the form of a program procedure. This model-interoperation approach contrasts with the approach of deriving spatial simulations purely from abstract statistical models, such as in the Cellular Potts Model (François and Glazier, 1992). μCell's main contribution is its approach to interoperation between signalling models and procedural models of cell components. The aim of μCell is to improve understanding of the mediating role of cell biochemistry to spatial behaviour.
This paper introduces μCell and, as an example, discusses the modelling and simulation of flagella-based chemotaxis, a form of cell motility, in E. coli bacteria. μCell is the result of interdisciplinary research undertaken by a group of six final-year undergraduate students in the Department of Computer Science at the University of Warwick during 2007/2008 for the fulfilment of the final-year group project requirement for the MEng Computer Science course[2]. The students involved chose to use this opportunity to learn about a different scientific discipline and do significant research rather than concentrate on software development.
The University of Warwick is a leading university in terms of interdisciplinary biological research, with the Warwick Systems Biology Centre[3], Warwick Complexity Complex[4], and the Molecular Organisation and Assembly in Cells Centre[5]. Furthermore, the University of Warwick is a forerunner in undergraduate research with the Undergraduate Research Scholarship Scheme[6], and the Reinvention Centre for Undergraduate Research[7]and its journal[8], with opportunities for research-based projects in many undergraduate courses.
The project was supervised by Sara Kalvala, a computer scientist with research interests in the field of computational biology and conducted under the advisement of David Whitworth, a biologist involved in systems biology research. Central aims of the MEng group project are to:
-
promote experience in collaborative team work on substantial projects,
-
improve communication skills,
-
improve skills in team and project management, raising awareness of the issues involved in group projects,
-
gain experience in co-authoring a sizeable project report.
This paper is structured as follows. The Materials and Methods section describes the materials and methods, including the computer science methods and software development techniques employed in developing μCell, the key components of the μCell software, and the example models used to simulate chemotaxis. The results section describes the results of chemotaxis simulations in μCell and the learning outcomes of the project. We then give a discussion of related work, followed by further work and conclusions.
Materials and Methods
The Development of μCell
The team had a diverse range of software development and computer science skills that were employed in developing μCell, incorporating 2D and 3D graphical programming, XML data handling, parsing and handling syntax trees, object-oriented data patterns, and functional programming. The biological aspect of the project presented the team with a significant challenge due to insufficient understanding of the problem domain as no member of the team had a background in biology. The agile software development methodology (in particular extreme programming (XP) (Beck 1999)) was therefore employed to minimise the risk of not satisfying the requirements of the software. Agile methodologies such as XP promote multiple, short development iterations throughout a project with frequent evaluation phases. Change is facilitated as the team develops a deeper conceptual understanding of the task, which is informed by their own development and exploration.
Prototyping was used to learn the biological background and explore the core component requirements of μCell. Learning was enhanced through enrolment in a level M computational biology module[9] by all team members.
The foundational components of the μCell program (such as the data structures, data parser, formula parser, and simulator) were developed using the test-driven development approach, where the design process for a program component's functionality is followed by development of test code, giving a procedural specification for the intended functionality of a component prior to implementation. Extensive tests form a basis for testing the functional equivalence of code between development iterations, thus enabling safe refactoring and reimplementation.
The team used the object-oriented language C# for its speed and cross-platform capabilities. The team employed standard collaborative development tools such as Subversion for source code and documentation version control, BaseCamp project management for online collaborative design and decision making, and the Eventum bug tracking system for collecting bugs and assigning members to resolve the issues. The Visual Studio integrated development environment and the MonoDevelop environment were used as development platforms.
μCell overview
The μCell software comprises three main components: the editor, simulator, and analyser. Included in this section are screenshots of μCell. The software and manual can be downloaded from the μCell Wiki[10].
Editor
The editor provides an interface for the construction and configuration of cell models and experiments. An experiment consists of a set of cell models, called cell definitions, and a set of simulations. Simulations consist of an environment, a configuration of cells instantiated from cell models, and various parameters governing data capture for analysis. Cell models are defined in terms of signalling pathways that model the biochemical reactions within a cell, and in terms of a set of predefined cell component models. These procedural cell component models may have parameters but are atomic: the user cannot deconstruct them or modify them directly.
Signalling pathways are constructed in the cell definition editor by drawing graphs of reactions and molecules (called species) (Figure 1) where reaction rates are described in mass action form. Pathways can be imported and exported using the Systems Biology Markup Language (SBML) (Huckaet al., 2003) standard. The procedural cell component models can be integrated into a signalling pathway as nodes in the pathway graph where input and output points of the components are linked to species. Thus, the procedural cell component models can be placed in the signalling pathway and can affect changes or be affected.
Simulations are constructed by configuration of an initial state, including the size and shape of the environment (box, tube, petri-dish, or sphere/orb) (Figure 2), spatial arrangement of cells, staining of cells (as is standard practice in real-world experimental biology) (Figure 3), concentration gradients (Figure 4), and other global parameters regarding the environment or simulation. For statistical analysis and data logging, time-series can be defined in terms of formulae associated with the simulation to be calculated at user-defined time intervals (Figure 5).
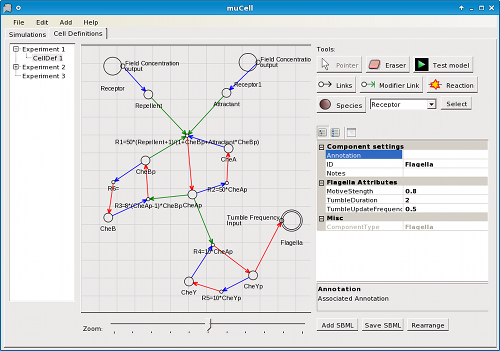
Figure 1. μCell cell model editor - Editing an imported SBML model.
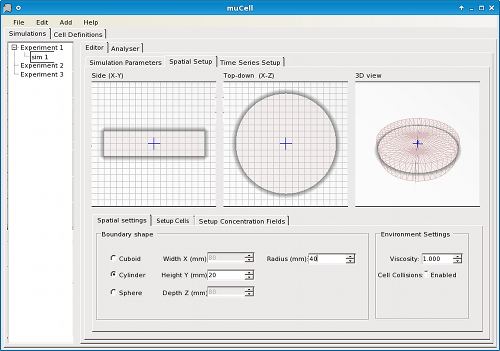
Figure 2. μCell spatial environment editor - Define the size and shape of the simulation environment.
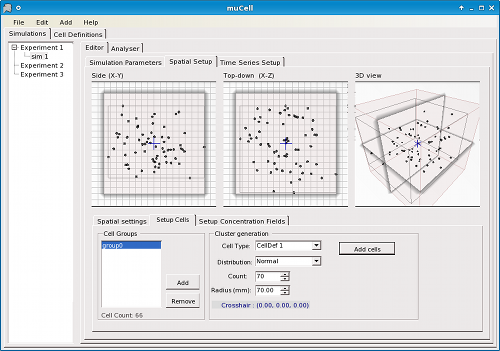
Figure 3. μCell spatial editor for cells - Defining initial cell placement.
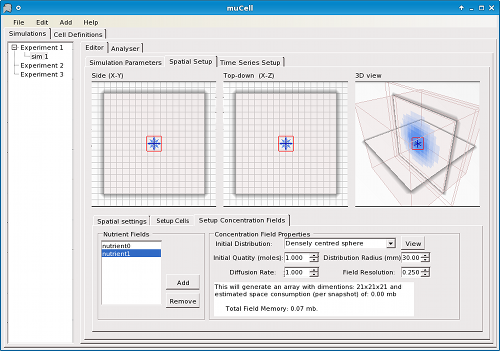
Figure 4. μCell concentration gradients editor - Defining concentration fields and diffusion.
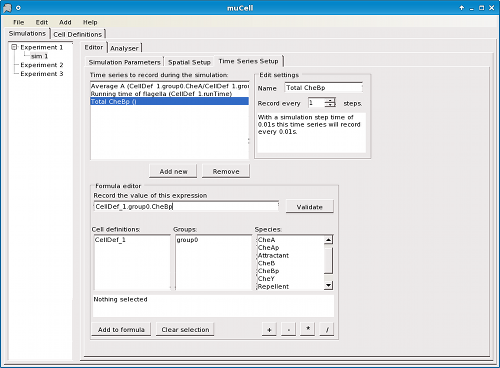
Figure 5. μCell time series editor - Defining formulae for time-series data to be generated by a simulation.
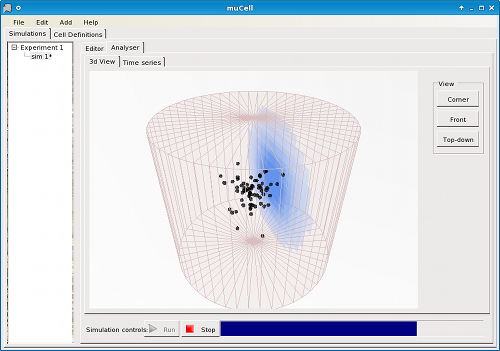
Figure 6. μCell Analyser - 3D simulation environment shows the spatial position of cells. A 2D planar view of the concentration scans over the environment.
Simulator
The simulator runs a simulation from a given experiment. Cells are instantiated from cell definitions (models) each having their own set of internal chemical concentrations used in the signalling pathway, spatial properties (such as velocity and orientation), and parameterised components belonging to the cell described by the procedural models.
Signalling pathways are simulated using finite difference techniques such as the Runge-Kutta and Euler methods for the approximation of differential equations. Approximations are computed iteratively using the time step interval specified in the simulation configuration.
Whilst pathways are simulated via approximations of differential equations, the procedural models of cell components are hard-coded as C# classes with "step" methods run at each iteration of the simulation which may interact with local chemical constituents, either within the cell, or in the environment at the current spatial position. Procedural models can affect other parameters of the cell such as spatial properties.
An additional spatial simulator, which resembles a classic physics simulator, maintains cell locations, collisions, and boundaries within a continuous Cartesian space, as opposed to quantising locations into a grid as in cellular automata. In the current simulator, cells reaching a boundary are simply deflected without loss of energy. By default the cells are represented as points in the space but can be given a size and mass via the "cell body" component for simulating collisions.
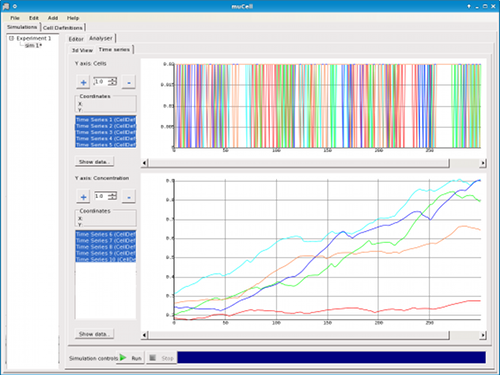
Figure 7. μCell Analyser - Plots showing concentration of attractant during chemotaxis and run lengths for 5 sampled cells.
Analyser
The analyser provides an interface for viewing data collected during and after a simulation, and provides feedback on simulation progress. The analyser provides a 3D representation of the spatial environment and the cells within it (Figure 6). Concentration gradients are shown via a 2D planar slice that moves through the environment showing a coloured representation of the concentration in that plane where a lighter colouring corresponds to a low molar concentration and a darker colouring corresponds to a high concentration.
The analyser also provides access to time-series data generated during simulation in the form of plots (Figure 7). The raw numerical data for the plots can be viewed and exported to CSV format for importing into other software, spreadsheets, or analysis tools.
Modelling and Simulating Chemotaxis
The following section explains the features of μCell that allow the modelling and simulation of complex behaviours in cells that bring together signalling pathways and cell components. In particular, we explain how μCell can be used for the modelling and simulation of flagella-based chemotaxis in E. coli towards an arbitrary attractant through a biased random walk.
In the simulation, the random walk effect of the chemotaxis is induced by the interoperation of signalling pathway models with procedural models of receptor and flagella, and simulations of concentration gradients. The simulated random walk behaviour of chemotaxis is not encoded explicitly in any of the models but is an emerging behaviour of the system.
Introduction to Chemotaxis
Chemotaxis is a form of cell motility that is promoted by chemical concentrations in an environment. Chemicals promoting chemotaxis may act as attractants or repellents, affecting the average directional movement of a cell. Remarkably, chemotaxis allows a cell to move up an attractant concentration gradient (or down a repellent concentration gradient) despite the fact that a cell is too small to measure a concentration gradient in space. The cell overcomes its size limitation by measuring the concentration gradient temporally (Sourjij, 2004).
There are many modes of transport for a cell performing chemotaxis. Here we focus on chemotaxis facilitated by flagella. Motile force is provided by a bundle of flagella which rotate either clockwise or anticlockwise. Anticlockwise rotation of the flagella produces a force directed towards the centre of the cell, propelling the cell forwards in a straight line called a run. Clockwise rotation of the flagella produces a force directed away from the centre of the cell resulting in the cell tumbling randomly on the spot. The cell alternates between the two phases of either tumbling or running with some tumble probability affecting the tumble frequency. A temporal increase in attractant, or decrease in repellent, reduces the flagella tumble probability, thus reducing the tumble frequency, resulting in longer runs when moving up a concentration gradient of attractant or down a concentration gradient of repellent. Conversely a decrease in attractant, or an increase in repellent, increases the tumble probability, therefore runs become shorter (Berg and Brown, 1972). Thus, the cell performs a random walk with a bias towards a high concentration of attractants and a low concentration of repellents.
The cell adapts its internal biochemistry to the new level of attractants in the environment as it moves in a straight line so that the cell converges to a stable tumble frequency in both extremely high and extremely low levels of concentration. In effect, the tumble frequency is independent of the magnitude of concentration; it is a temporal gradient of concentration that is required to stimulate the cell to alter its tumble frequency.
The link between chemotaxis and flagellar motion has been the subject of much interest over the last 30 years, and E. coli has been a very commonly used model for the study of this mechanism. The signalling pathway for sensing is well understood (Macnab, 1996), however 'much remains to be understood’ about the flagellar motor (Sowa and Berry, 2008). The extensive knowledge of E. coli motility has been derived from wet experiments and biophysical modelling; a spatial simulation linking the flagellar motor’s effect with the signalling pathways could be a useful addition to the arsenal of tools available to bacteriologists.
Our chemotaxis model is derived from a simple metabolic network of just three molecules and procedural models of receptors and flagella. The procedural model for a receptor is extremely simple in μCell, taking a sample of a specified molecule's concentration at the cell's current spatial position. The model of a receptor thus resembles the very entry point of a molecule into a cell—there are no methylation bindings for controlling reception in the model. Concentration gradients and diffusion are simulated in μCell by a lower resolution approximation of the spatial environment. We describe the method for simulating concentrations first, followed by the signalling pathway used in the chemotaxis model, and finally the flagella bundle model.
Simulating concentration gradients and diffusion
μCell discretises the concentration gradients in the spatial environment by approximating the environment as a set of discrete cubes of uniform volume that describe a uniform molar quantity of some molecule. The size of the cubes, hence the resolution of the approximation, can be configured in the simulation parameters to suit available system resources; higher precision approximations result in greater memory usage. A mapping from points in the continuous spatial environment to corresponding discrete cubes returns the concentration of a molecule at a point in the environment. Diffusion is simulated across each face of a cube at a constant diffusion rate specified by the user.
The initial distribution of a concentration can be specified either uniformly or as a densely centred sphere that approximates an initial diffusion from a fixed point. An initial quantity is set by the user specifying the total concentration of the molecule that will be added to the environment.
The discretisation process poses a problem for chemotaxis as it introduces sudden, sharp changes in concentration between cubes and uniform concentrations inside a cube. Chemotaxis relies on continuous changes in the concentration gradient. The discretisation problem is overcome by interpolation using Gaussian weighting (see Appendix A).
Signalling pathway for Chemotaxis
The signalling pathway we use for the chemotaxis model is derived from the known chemotaxis pathway of E. coli with some simplifications; Figure 8 shows the pathway taken from the KEGG database. We focus on the molecules: CheA, CheB, CheR, CheW, CheY, and CheZ. Attractants and repellents are bound to specific transmembrane receptors (chemoreceptors) which are coupled with a scaffolding protein (CheW) and a histidine kinase (CheA) to form complexes within the cell. CheA phosphorylates itself modifying the rate at which a messenger protein (CheY) is phosphorylated. The phosphorylated form of CheY (CheYp) modulates the flagella bundle's tumble probability. Binding of attractants to receptors decreases the rate of CheA phosphorylation, which decreases phosphorylation of CheY, resulting in a lower probability of tumbling, explaining why higher concentrations of attractants produce longer runs. The pathway adapts to high and low concentrations so that the chemotaxis behaviour is solely dependent on temporal changes in concentrations, independent of magnitude.
Adaption to a concentration's magnitude occurs via methylation of the chemoreceptors controlled by CheBp which demethylates the receptor and CheR which methylates the receptor. CheAp inversely promotes the phosphorylation of CheB. In low concentrations of attractant and high concentrations of repellent the amount of CheAp is high resulting in less CheB phosphorylation thus less demethylation of the receptor. This increases the methylation effect of CheR, and therefore the amount of CheA phosphorylation, which increases the phosphorylation of CheY. Thus, in lower concentrations of attractant, the tumble probability increases. CheY is dephosphorylated rapidly by CheZ. For more details see (Alon et al., 1999; Falke et al., 1997).
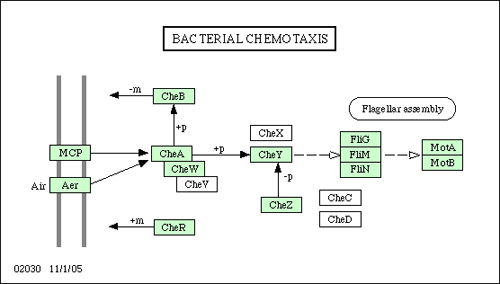
Figure 8. Pathway for bacterial chemotaxis in E. coli taken from the KEGG database (Kanehisa & Goto 2000) (KEGG Retrieved December 2008).
We use a simplified version of this pathway where methylation through CheR is removed by using only CheB to directly affect adaption. Because the receptor models are extremely simple without adaption through methylation, the methylation of receptors must be approximated within a single reaction which mediates the phosphorylation of CheA. Thus, the adaption process is simplified in our model to allow it to be more easily expressed in a single reaction. The implication of this simplification is that adaption is faster acting and has a greater effect as it is not mediated through a fine balance of methylation by CheR and demethylation by CheBp. Additionally, we eliminate the scaffold molecule CheW, due to the simplified receptors, and the promoter of CheY dephosphorylation, CheZ, as it is a constant that can be expressed in the reaction. These pathway simplifications fit our simplified receptor model, making it quicker to simulate than a full adaption process.
Figure 9 shows the chemotaxis pathway defined in μCell. Reaction rates are defined in μCell in mass action form. For the chemotaxis pathway the reactions are:

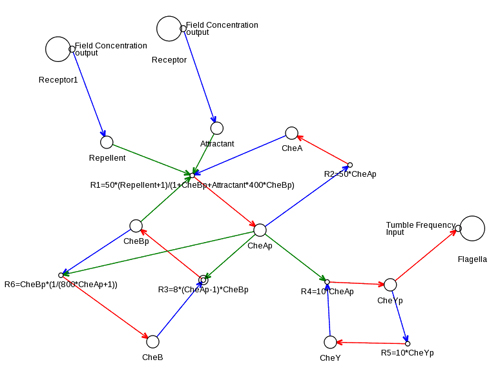
Figure 9. The chemotaxis pathway in the μCell cell definition editor.
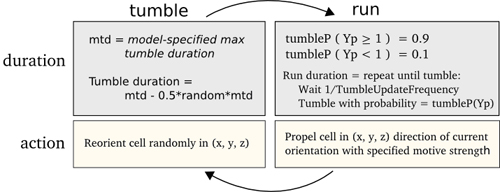
Figure 10. Informal description of the tumble and run behaviours in the flagella bundle model.
Modelling Flagella
As described above, the tumble probability of a cell is controlled by the effects of the phosphorylated CheY molecule on the flagella bundle. Figure 10 informally describes the procedural model of the flagella bundle. The flagella bundle model consists of two alternating states: running and tumbling. Each state has a duration of time spent performing the action of that state.The tumble duration is determined by the maximum tumble duration specified in the flagella parameters minus a random scaling, between 0 and 0.5, of the maximum tumble duration. During tumbling, the cell re-orients itself randomly. The run duration is a multiple of the reciprocal of the tumble update frequency, i.e. tumble update time (specified in the cell definition editor), which acts as a minimal unit of running time. The concentration of CheYp is tested in each iteration of the run state and changes the likelihood of switching back to tumble. When CheYp is high, the likelihood of switching is high (0.9), when it is low the likelihood of switching is low (0.1). During a run the cell is propelled forwards in the current orientation.
This model is hard-coded in C# into μCell. In future, we hope to provide an abstract procedural modelling language for modifying and defining new components (see Further Work).
Chemotaxis Experiments
In order to test whether μCell was able to capture the link between models for flagella and receptors and a known signalling pathway controlling chemotaxis, we carried out simulated experiments testing the operation of the models.
Experiment 1
The first experiment tests the average run duration of chemotaxis in the presence of an attractant. A chemotaxis-enabled cell was placed in an environment with an attractant concentration gradient; the average run duration was measured and compared with the average run duration for a control cell in an environment where no gradient was present. The tumble durations were also measured, with the understanding that they would be approximately the same in both cases, as, in our model, the tumble duration is independent of the attractant concentration.
The spatial environment of the experiment is a cuboid of size 80x80x 80mm3. Into the environment we placed a cell whose model contains the chemotaxis pathway described previously, a single attractant receptor model, and a flagella bundle model, configured with the default motive strength 0.8, tumble update frequency 0.5, and maximum tumble duration 2. The cell was placed near the edge of the environment at co-ordinates (-25, 0, 0), 15mm from the environment boundary and 25mm from the centre of the environment. In the control test there was no attractant present. In the main test, we specified a densely centred sphere of attractant concentrated at the centre of the environment, with a radius of 30mm and a central concentration of 10.0 micromoles in total. This is the equivalent to injecting a 10.0 micro-molar attractant concentration at the centre of the environment and allowing it to evenly diffuse before the start of the experiment up to a radius of 30mm from the centre in each direction. The constant rate of diffusion we used is low, at 0.001.
The experiment was run for 400s with a temporal resolution of 0.05s. The control test and main gradient test were repeated 5 times each with 5 cells tracked each time. The mean run duration of cells was measured. The results are shown in Experiment 1 below.
Experiment 2
The second experiment tested whether the average run durations of a cell eventually decrease when a high concentration is reached, thus allowing cells to congregate at the highest concentration of an attractant and remain in the vicinity. The second experiment also tested whether cells actually move up the concentration gradient towards the higher concentration of attractant.
The spatial setup was the same as the first experiment, although here we used 100 cells uniformly distributed in the spatial environment. In the control test we placed no attractant and in the main test we again added a 30mm radius densely centred sphere concentration gradient of 10 micromoles in total. The constant rate of diffusion used was low at 0.001. The experiment was run for 800s with a resolution of 0.05s and observed 3 times.
The average attractant within the cell population was measured during the experiment, and we hypothesized that it would steadily increase as the cells congregate in the centre, but that the first derivative of the cell population curve would eventually decrease as cells reach the relatively stable region of the high concentration. The results are shown in Experiment 2 below.
Results
Described in this section are the results from the two chemotaxis experiments in μCell and a brief note on the results of the project in terms of the μCell tool and the learning outcomes for the team.
Chemotaxis Experiments
The general behaviour observed in these simulations matched the experimental behaviour of E. coli chemotaxis reported in the literature.
Experiment 1
The results for the first experiment are tabulated in Figure 11. When cells were placed in the environment with an increasing concentration gradient towards the centre, the mean run duration increased to just over twice that of the control group with no attractant present. This matches the expected behaviour of cells performing chemotaxis towards an attractant. The mean maximum run duration was much higher in the main experiment than in the control experiment.
As predicted, the mean tumble time was roughly the same (up to 2 decimal places) in the control and the main experiment, with a very slight difference in the mean deviation, as the tumble duration is independent of the attractant concentration. The maximum tumble durations are identical, as they are fixed in the flagella model at 2s as stated in the experiment method (the 0.05 extra represents one extra time step that can elapse before switching state).
|
|
Control |
Gradient |
|
|
Concentration (moles x10-6) |
0 |
10 densley centred sphere |
|
|
Mean tumble duration (s) |
1.52 +/- 2.77 |
1.52 +/- 2.73 |
|
|
Mean run duration (s) |
2.25 +/- 6.25 |
4.55 +/- 0.42 |
|
|
Max run duration (s) |
5.82 +/- 1.10 |
29.68 +/- 6.76 |
|
|
Max tumble duration (s) |
2.05 +/- 1.33 |
2.05 +/- 1.33 |
Figure 11. Experiment 1 results (to 2 decimal place) +/- mean deviation
Comparing Results of Experiment 1 to Real Experimental Data
We compared the results from experiment 1 with real-world experimental data for chemotaxis of an E. coli "wild type" in a capillary tube with a concentration gradient of 10 micro moles of serine from (Rojdestvenski, 2003):
|
|
Control |
Gradient |
|
|
Concentration (moles x10-6) |
0 |
10 |
|
|
Mean run duration (s) |
0.83 +/- 0.88 |
1.67 +/- 2.56 |
Although these measurements differ from our simulation results, both exhibit the same behaviour of run duration roughly doubling in the presence of a gradient. In fact, we can use experimental data such as this to adjust the model parameters. In this case, the tumble update frequency can be scaled according to the magnitude difference between the μCell results and the real-world results. Using a tumble update frequency of 1.355 instead of the previous 0.5, μCell measures the following:
|
|
Control |
Gradient |
|
|
Concentration (moles x10-6) |
0 |
10 |
|
|
Mean run duration (s) |
0.83 +/- 1.44 |
1.74 +/- 0.33 |
Note, however, that the average deviation is very different between the experimental data and the data collected from μCell. We conjecture that this is a result of the approximated adaption pathway in our pathway model for chemotaxis. The approximated adaption pathway results in overly quick adaption to the magnitude of the relevant concentration. Therefore, the mean deviation of a cell's tumble duration when in a gradient is lower than expected as the pathway adapts more quickly to a higher concentration. The probability weightings of the flagella model may also require further tuning. Further collaboration with biologists would be needed to tune the models based on results from real-world, in vivo experiments.
Experiment 2
Figure 12 shows that in our simulation the mean attractant over all cells, in all three experiments, increased in the presence of a concentration gradient and the first derivative of the curve decreased as the cells reached the area of greatest nutrient concentration. The curve is irregular at its limit due to tumbling and short runs resulting in occasional forays into areas of slightly lower concentration.
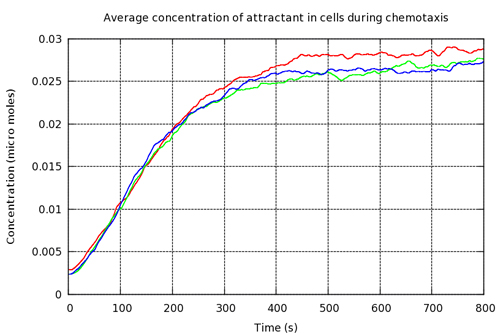
Figure 12. Mean attractant across all cells increased as they moved up the concentration gradient, eventually reaching a plateau as cells converge at the area of highest concentration.
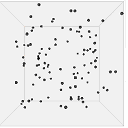
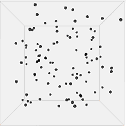
Figure 13 shows one set of visual results from the 3D spatial environment for the first 400 seconds of the simulated time. In the control situation (Figure 13(b)), the cells wander randomly with no attractant to promote directed movement. Conversely, in the presence of a concentration gradient (Figure 13(a)), almost all the cells converge in the high concentration area in the environment's centre. There are a few cells that have not reached the centre due to the fact that chemotaxis, even though it is a biased walk, is a random walk and therefore can be unsuccessful for a long time.
| 0s |  |
 |
| 100s | 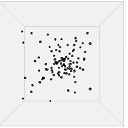 |
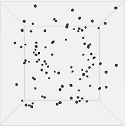 |
| 200s | 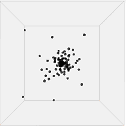 |
 |
| 300s |  |
 |
| 400s |  |
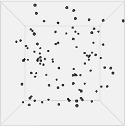 |
| (a). Cells in 10 micromoles of centre weighted attractant presenting chemotaxis towards the high concentration area. | (b). Cells in 0.0 moles of attractant presenting no chemotaxis. |
Figure 13. Screen captures of the 3D spatial environment shown from the front during simulation at approximately 100 second intervals.
The µCell software
The μCell tool developed during the project was successful in its goal to be practical and usable for the modelling and simulation of cell behaviour in spatial environments. The tool is easy to use and may also be useful as a teaching tool in biology. The tool is now freely available as open-source software and has a public code repository and 'Wiki’[11]. A video is available showing μCell being used in a chemotaxis experiment[12]. Development of the tool has continued, albeit slowly, since the formal end of the project, and we expect to collaborate further with biologists to improve its usefulness. The μCell tool will be discussed in the context of related work in the next section.
Learning outcomes
The team's study of the undergraduate computational biology course and the work on the μCell project were mutually beneficial, promoting research as learning, and providing domain-specific knowledge for interdisciplinary research. Biological insights were developed parallel to the computer science skills acquired and developed during the project, including: collaborative software development, use of the C# language, and the use of tools for software development, version control, and bug tracking. Additionally, skills in project management, communication, writing, and collaboration were learnt and reinforced.
The team thoroughly enjoyed the project and benefited greatly from being involved in a different area of scientific research from computer science. The team was exposed to biological research, and more generally to methodologies and common practices in another scientific field. Group members gained an understanding of common cell processes, biochemistry, signalling pathways, diffusion mechanics, flagella mechanics, SBML, and of the issues in computational modelling and simulation of cells.
The project was deemed to have satisfied the requirements for the MEng group project and all students received a first class grade for the work.
Related Work
There are numerous tools that provide modelling and cell design abilities and/or simulation features to aid research in biology. A few projects that share some commonality with μCell will be discussed here.
The modelling and simulation of signalling pathways can be performed with many existing tools; the SBML website provides an extensive list of software supporting SBML models[13]. μCell does not claim to improve upon these tools in terms of speed of simulation or SBML-support but is novel, as far the authors are aware, in providing a tool for combining pathway models with pre-defined models of cell components for the simulation of spatial behaviour.
Virtual Cell, an advanced tool for cell simulations based on layers of models, performs complex spatial simulations of compartments and membranes within a single cell derived from real geometric data (Moraru et al., 2008). The spatial simulations within Virtual Cell are at a finer granularity than μCell's spatial simulations, which are at the multi-cellular level.
The BioSPICE open source framework for modelling and simulation in systems biology is an extendable framework with a multitude of pluggable modules that can be configured to interoperate (Kumar & Feidler, 2003). Systems Biology Workbench (SBW) is a grand effort to combine the best tools from all aspects of biological modelling into a single complete framework to enable interoperation between tools via standardisation (Hucka et al., 2001). The SBML standard is at the heart of this endeavour, originally introduced by the same organisation. BioSPICE and SBW are both formidable systems that are at times difficult to use, with a high barrier of entry for inexperienced users. μCell is more specific and succinct in its purposes and features, although the spirit of interoperation is shared at a smaller scale of model interoperation.
Another approach to modelling and simulation of spatial behaviours is through the programming of custom models, either through generalised modelling tools or programming languages. For example, NetLogo (Tisue & Wilensky, 2004) provides a generalised programmable modelling environment into which μCell-like models and simulations could be programmed. Other spatial models have been implemented using custom programming languages, such as the simulation of aggregation in Dictyostelium molds using a specially developed "cell" programming language (Agarwal, 1994). μCell provides specialised modelling and simulation tools for emerging behaviours of cells without requiring any programming experience from the user.
The CompuCell3D modelling environment provides 2D and 3D visualisations of various cell models (Cickovski et al., 2005), including cellular Potts model simulations of morphogenesis – the shaping of cells. CompuCell3D is also capable of simulating other processes, such as clustering, cell death, cell haptotaxis (the directional growth of cells), and chemotaxis. CompuCell3D uses custom modules written in C++ and Python, requiring programming expertise from the user and custom rule-bases to produce models and simulations, as opposed to providing pre-defined models and using lower-level biochemical dynamics to drive these processes as in μCell.
Spatial behaviours have been modelled and simulated using statistical models, such as extended Potts models (François & Glazier, 1992), whose behaviours are not mediated by signalling pathways as in μCell.
Further Work
There is a considerable amount of potential further work. A more extensive list with outlined research proposals for student projects is available on the μCell Wiki[14]. Here we briefly discuss a few areas of work.
Currently, the numerical accuracy of measurements within μCell is correct up to the definition of the models, but measurements taken from simulations may not match measurements from in vivo experiments. As suggested in Comparing Results of Experiment 1 to Real Experimental Data above, the parameters of models can be tweaked to give data that matches experimental data to an extent, but further collaboration with biologists is required if more realistic measurements are to be achieved. An example usage of μCell in research may be the modification of a signalling pathway due to a genetic fault and to test the effects of this modification on the emerging behaviours of the cell. For example, in E. coli chemotaxis, if CheY cannot phosphorylate quickly enough due to some unusual inhibition relation, will chemotaxis occur?
Currently, there are only procedural models for flagella bundles, receptors, and cell bodies (which facilitate cell collisions). This set should be extended to include components for modelling further processes such as cell growth, death, mitosis and cytokinesis (cell division), haptotaxis, and cell excretion. Furthermore, there should be tools for the user to develop their own procedural models of cell components. This may involve some form of synthetic language, or construction tool that should be sufficiently flexible to allow the definition of current and future models.
Additionally there are many improvements that can be made to μCell within current requirements to improve the user experience, such as more comprehensive error messages, improved saving and loading facilities, and the fixing of known bugs in the user interface.
Conclusions
This paper has introduced the μCell software tool for modelling and simulation of multi-cellular environments with a particular focus on the spatial behaviour of cells. The spatial behaviour of cells can be modelled in μCell via the interoperation of signalling pathway models (defined in the SBML data format) with pre-defined procedural models of cell components, such as flagella bundles. This approach to spatial simulations, controlled by signalling pathways, differs from stochastic approaches or cellular automata based approaches. The capabilities of this tool were demonstrated with two experiments showing μCell's ability to model and simulate E. coli chemotaxis. A user of μCell is not required to do any programming of their own as the procedural models of cell components are built-in and configurable.
At a meta-level, this paper has also presented a case study of interdisciplinary research at undergraduate level. The research project was undertaken by a group of six Level 4 computer science students at the University of Warwick. Their initial lack of biological knowledge required them to undertake their own learning, through courses, books, papers, and discussions with biologists in order to understand the problem domain. Thus, an outcome of this project, apart from the μCell software, has been the increase in the students’ scientific knowledge, and proliferation of skills in research.
It is our hope that the successful development of μCell and the outcomes described in this paper inspire further students to take on interdisciplinary research at both undergraduate and postgraduate level.
Acknowledgements
Many thanks are due to David Whitworth for all his help during the project. Thank you to all the biologists and biochemists who endured our many questions. Thanks also to the helpful comments of the anonymous referees.
Appendix A - Interpolation using Gaussian Weighting
The discretisation of the continuous simulation environment into cubes produces discontinuous changes in concentration between cubes and a constant concentration inside a cube. Chemotaxis relies on continuous changes in the concentration gradient thus the discontinuity and quiescence poses a significant problem for the chemotaxis simulation. The problem is overcome by interpolation using Gaussian weighting.
A co-ordinate (x, y, z) in the simulation environment has a mapping to a cube with co-ordinates (i, j, k) in the approximated concentration space. Each cube has 26 neighbours that are connected either by an edge, face, or vertex. The 27 cubes surrounding (i, j, k), including itself, are sampled, taking a weighted average of each where the weighting is based on the distance from the (x, y, z) to the centroid of each cube (illustrated in Figure 14 with 5 cubes as opposed to 27). A Gaussian function is applied to each distance to provide smooth interpolation:
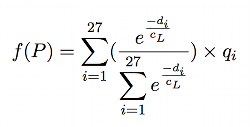
Where diis the distance from the point P to the centre of the ithcube centre in the array of 3x3x3 cubes surrounding point P, qiis the quantity in the ithcube, and finally cLis the length of the cube's side. Note that in general:
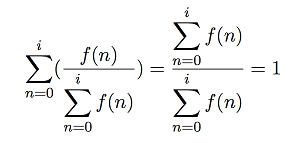
Hence, the weighting kernel will always sum to 1. For performance reasons, the nested summation is computed separately in an initial pass of the 27 cubes, stored, and then used in a second pass to avoid nested computation.
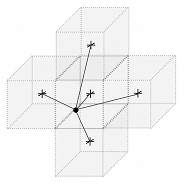
Figure 14. Nutrient field sampling interpolation scheme
Notes
[1] Author information will be available shortly.
[2] http://www2.warwick.ac.uk/fac/sci/dcs/teaching/modules/cs407
[3] http://go.warwick.ac.uk/systemsbiology
[4] http://go.warwick.ac.uk/complexity
[5] http://go.warwick.ac.uk/moac
[6] http://go.warwick.ac.uk/urss
[7] http://go.warwick.ac.uk/reinvention
[8] http://go.warwick.ac.uk/reinventionjournal
[9] http://www2.warwick.ac.uk/fac/sci/dcs/teaching/modules/cs904/
[10] http://github.com/dorchard/mucell/wikis/
[11] http://github.com/dorchard/mucell/wikis
[12] http://github.com/dorchard/mucell/wikis/screenshots-and-videos
[13] http://sbml.org/SBML_Software_Guide/SBML_Software_Summary
[14] http://github.com/dorchard/mucell/wikis/further-work
References
Agarwal, P. (1994), 'Simulation of aggregation in Dictyostelium using the cell programming language’, Computer Applications in the Biosciences, 6(10), 647-655
Alon, U., M. Surette, N. Barkai and S. Leibler (1999), 'Robustness in bacterial chemotaxis', Nature, 397(6715), 168-171
Beck, K. (1999), 'Embracing Change with Extreme Programming', Computer, 32(10), 70-77
Berg, H. C. and D. A. Brown (1972), 'Chemotaxis in Escherichia coli Analysed by Three-dimensional Tracking', Nature, 239(5374), 500-504
CellDesigner, Celldesigner website, http://www.celldesigner.org/, accessed December 2008
Cickovski, T. M., C. Huang, R. Chaturvedi, T. Glimm, H. G. E. Hentschel, M. S. Alber, J. A. Glazier, S. A. Newman and J. A. Izaguirre (2005), 'Compucell3d', IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB), 2, 273-288
Falke, J. J., R. B. Bass, S. L. Butler, S. A. Chervitz and M. A. Danielson (1997), 'The Two-component Signalling Pathway of Bacterial Chemotaxis: A Molecular View of Signal Transduction by Receptors, Kinases, and Adaptation Enzymes', Annual Review of Cell and Developmental Biology 13, 457-512
Hucka, M., A. Finney, H. Sauro, and H. Bolouri (2001), 'Introduction to the Systems Biology Workbench', JST ERATO Kitano Symbiotic Systems Project
Hucka, M., A. Finney, H. M. Sauro, H. Bolouri, J. C. Doyle, H. Kitano, et. al. (2003), 'The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models', Bioinformatics 19 (4), 524-531
Hynes, R. and A. Lander (1992), 'Contact and adhesive specificities in the associations, migrations, and targeting of cells and axons', Cell 68, 303-322
Kanehisa, M. and S. Goto (2000), 'KEGG: Kyoto Encyclopedia of Genes and Genomes', Nucl. Acids Res. 28 (1), 27-30
KEGG, Bacterial Chemotaxis Escherichia coli K-12 MG1655, http://www.genome.jp/dbget-bin/show pathway?eco02030, accessed December 2008
Kitano, H. (2002), 'Systems Biology: A Brief Overview', Science 295 (5560), 1662-1664
Kumar, S. P. and J. C. Feidler (2003), 'BioSPICE: a computational infrastructure for integrative biology', OMICS: A Journal of Integrative Biology, 7 (3), 223-225
Macnab R. M. (1996), Flagella and motility p123-145. in Escherichia coli and Salmonella: Cellular and Molecular Biology, F. C. Neidhart et al (eds.), 2nded, ASM Press, Washington, DC
Moraru, I., J. Schaff, B. Slepchenko, M. Blinov, F. Morgan, A. Lakshminarayana, F. Gao, Y. Li, and L. Loew (2008), 'Virtual cell modelling and simulation software environment', Systems Biology, IET 2 (5), 352-362
Rojdestvenski, I. (2003), 'Metabolic pathways in three dimensions’, Bioinformatics19 (18), 2436-2441
Sourjij, V. (2004), 'Receptor clustering and signal processing in E. coli chemotaxis', TRENDS in Microbiology 12
Sowa Y and R. M. Berry (2008), 'Bacterial Flagellar Motor’, Quarterly Review of Biophysics, 41, 103-132
Tisue, S. and U. Wilensky (2004), 'NetLogo: A simple environment for modelling complexity’, International Conference on Complex Systems (ICCS 2004), Boston, MA, pp. 16-21
To cite this paper please use the following details: Orchard, D., Gover, J., Herrington, L., Lohr, J., Stead, D., Young, C. and Kalvala, S. (2009), 'muCell - Interdisciplinary Research in Modelling and Simulation of Cell Spatial Behaviour', Reinvention: a Journal of Undergraduate Research, Volume 2, Issue 1, http://www2.warwick.ac.uk/go/reinventionjournal/issues/volume2issue1/orchard Date accessed [insert date]. If you cite this article or use it in any teaching or other related activities please let us know by e-mailing us at Reinventionjournal at warwick dot ac dot uk.
© Reinvention: a Journal of Undergraduate Research (2009). Full copyright remains with the author.