Assembly & Dynamics of Macromolecular Complexes from Bacterial Pathogens
Principal Supervisor: Professor Phill Stansfeld
Secondary Supervisor(s): Depending on the project there are a number of colleagues who would be most appropriate for co-supervision, including but not limited to David Roper, Allister Crow, Seamus Holden, Liz Fullam, Chris Dowson, Alex Cameron, Greg Challis (Warwick), Andrew Lovering, Tim Knowles (Birmingham), Roslin Bill (Aston)
University of Registration: University of Warwick
BBSRC Research Themes:
- Understanding the Rules of Life (Microbiology, Structural Biology)
- Integrated Understanding of Health (Pharmaceuticals)
Apply now!
Deadline: 23 May, 2024
Project Outline
The development of the protein-folding software, AlphaFold2, has enabled the computational determination of over 130 million monomeric protein structures. One can also use this method to model both homo- and heteromeric complexes. This is especially true for bacterial proteins, where organism and operon-based information may be used to provide a blueprint for how AlphaFold may assemble the protein structure and/or complex. An example of one of our computer-modelled complexes is shown below. This is the target for beta-lactam inhibitors, e.g. penicillin.
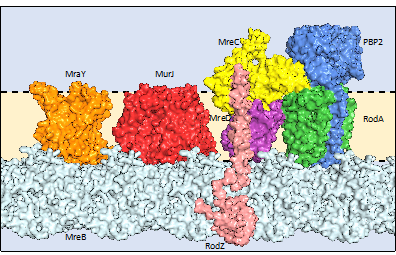
One quarter of the 130 million folded protein structures are expressed within lipid membranes. In bacteria, these proteins fullfill key tasks of, e.g., selective uptake of nutrients, processing of secreted proteins or expulsion of toxic compounds, such as antibiotics; and therefore form current and future targets for novel antimicrobials through inhibition of these processes.
The overall aim of this proposal is to provide a structural and dynamic description of bacterial complexes that form within the cell envelope, embedded within lipid membranes; the front-line to a bacterium’s defence.
By understanding the three-dimensional details of how these complexes form and move, we have a better grasp of the fundamental processes performed by these proteins. This therefore provides an improved understanding of how one can develop novel antimicrobial inhibitors.
References
- Ashraf KU, Nygaard R, Vickery ON, Erramilli SK, Herrera CM, McConville TH … Trent S, Stansfeld PJ, Mancia F. Structural basis of Lipopolysaccharide Maturation by the WaaL O-Antigen Ligase. Nature 2022. 604:371-376.
- Maloney FP, Kuklewicz J, Corey RA, Bi Y, Ho R, Mateusiak L, Pardon E, Steyaert J, Stansfeld PJ, Zimmer J. Structure, substrate-recognition, and initiation of hyaluronan synthase. Nature 2022; 604:195-201
- Oluwole AO,Corey RA,Brown CM, Hernández-Rocamora VM, Stansfeld PJ, Vollmer W, Bolla JR, Robinson CV. Peptidoglycan biosynthesis is driven by lipid transfer along enzyme-substrate affinity gradients. Nature Comms.
- Song W, Corey RA, Ansell TB, Cassidy CK, Horrell MR, Duncan AL, Stansfeld PJ, Sansom MSP. PyLipID: A Python Package for Analysis of Protein-Lipid Interactions from Molecular Dynamics Simulations. JCTC 2022;18:1188-1201.
- Corey RA, Song W, Duncan AL, Ansell TB, Sansom MSP, Stansfeld PJ. Identification and assessment of cardiolipin interactions with E. coli inner membrane proteins. Science Adv. 2021 7:eabh2217
- Vickery ON, Stansfeld PJ. CG2AT2: An Enhanced Fragment-based approach for Serial Multi-scale Molecular Dynamics simulations. JCTC 2021.
- Fiorentino F, Sauer JB, Qiu XY, Corey RA, Cassidy CK, Mynors-Wallis B, Mehmood S, Bolla JR, Stansfeld PJ, Robinson CV. Ligand induced conformational dynamics of the LPS translocon LptDE. Nature Chemical Biology 2021
Techniques
- AlphaFold2-based protein folding
- Molecular Dynamics (MD) simulations
- Structural Bioinformatics
- Python-based programming
- Ligand Docking
- Structure-based Drug Design