
Research
Machine-learning-enabled discovery of dynamics at interfaces
The past and the future of chemistry involve extreme conditions. Extreme conditions mean extremely high or low pressures and temperatures and extreme radiation and magnetic fields that are not easily realized on modern-day Earth. In the past, the molecules of life (proteins and nucleobases) were forged at a time when the world was extremely hot and no ozone layer was present to protect Earth’s surface from harmful intense UV radiation. Extreme conditions provided the driving force to create molecules that could resist those conditions and that formed the building blocks of our highly resilient genetic code. In the future, the technology and innovation base that will equip our fight against climate change and that will drive the transition to clean and abundant energy and food, crucially relies on exposing molecules to extreme conditions that are not naturally realised. By exposing molecules to extreme intensities of light, electric fields and magnetic fields, we can fundamentally change their behaviour. Molecular simulation and chemical informatics play a crucial role in enabling the study of chemistry under extreme conditions. The emergence of machine learning methods in the chemical and physical sciences has the potential to fundamentally transform our ability to predict and simulate chemical processes far away from equilibrium.
Our Research Vision
is to develop and apply new simulation methods and prediction tools that fuse machine learning (ML) and data-driven approaches with quantum mechanical (QM) electronic structure and dynamical simulation methods. Doing so will remove existing bottlenecks in simulation capabilities that limit the length and time scales and the complexity of chemical simulations. More importantly, hybrid ML/QM methods enable fundamentally new prediction approaches that defy the conventional structure-to-property paradigm that underpins modern chemical studies. The Maurer group applies newly developed ML-enabled research tools in the areas of high-throughput molecular design for organic electronics and functional interfaces, as well as the study of chemical dynamics triggered by extremely short or intense light and electromagnetic fields.
Current Research Topics in the Group
Novel Machine-Learning Representations in Electronic Structure Theory
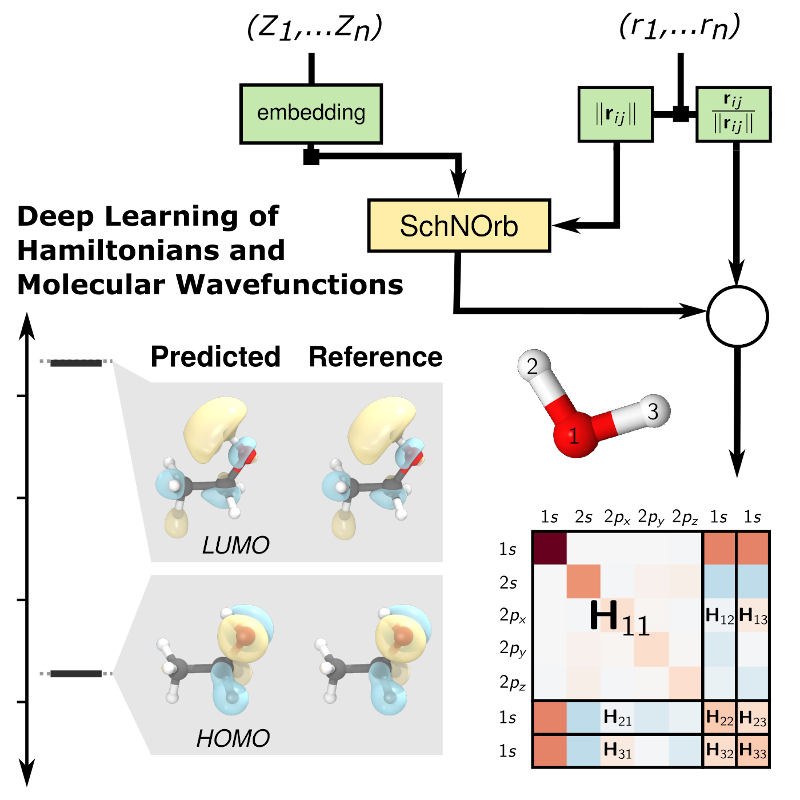
Collaborations:
-
Prof. Klaus-Robert Müller, TU Berlin
- Prof. James Kermode, University of Warwick
- Prof. Christoph Orthner, University of British Columbia
Selected Publications:
- T. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller, and R. J. Maurer, “Deep learning of quantum chemistry to predict the molecular wavefunction and more”, Nature Commun. 10, 5024 (2019)
- M. Gastegger, A. McSloy, M. Luya, K. T. Schuett, R. J. Maurer, "A deep neural network for molecular wave functions in quasi-atomic minimal basis representations", J. Chem. Phys. 154, 044123 (2020)
- J. Westermayr, R. J. Maurer, "Physically inspired deep learning of molecular excitations and photoemission spectra", Chem. Sci. 12, 10755-10764 (2021).
- L. Zhang et al. "Equivariant analytical mapping of first principles Hamiltonians to accurate and transferable materials models", npj Computational Materials 8, 158 (2022)
- J. Westermayr, Joe Gilkes, Rhyan Barrett, R. J. Maurer, "High-throughput property-driven generative design of functional organic molecules", Nature Comput. Sci. 3, 139–148 (2023)
Light- and Electron-Driven Dynamics at Surfaces and Nanostructured Interfaces

Collaborations:
-
Prof. Bin Jiang, University of Science and Technology, Hefei, China
- Prof. Alec Wodtke, Max Planck Institute for Multidisciplinary Sciences, Goettingen, Germany
- Prof. Michael Thoss, University of Freiburg, Germany
Explicit atomistic simulation of the dynamics of molecular reactions can help to gain valuable understanding of processes relevant to catalysis, surface chemistry, and surface scattering. Molecular dynamics on metal surfaces are especially interesting due to a variety of quantum and nonadiabatic effects that determine the qualitative and quantitative outcome of reactions. Such effects include non-adiabatic energy transfer between adsorbate motion and substrate electron-hole pair excitations leading to vibrational cooling of adsorbate motion or quantum nuclear effects that facilitate barrier crossings via tunnelling. Our current efforts are in developing methodology to include the above quantum effects in the study of non-equilibrium processes on surfaces.
Selected Publications:
- O. Douglas-Gallardo, C. Box, R. J. Maurer, "Plasmonic enhancement of molecular hydrogen dissociation on metallic magnesium nanoclusters", Nanoscale 13, 11058-11068 (2021)
- C. L. Box, Y. Zhang, R. Yin, B. Jiang, R. J. Maurer, "Determining the effect of hot electron dissipation on molecular scattering experiments at metal surfaces", JACS Au 1, 164-173 (2021)
- J. Gardner, D. Corken, S. J. Janke, S. Habershon, R. J. Maurer, "Efficient implementation and performance analysis of the independent electron surface hopping method for dynamics at metal surfaces", J. Chem. Phys. 158, 064101 (2023)
- C. L. Box, W. G. Stark, R. J. Maurer, "Ab initio calculation of electron-phonon linewidths and molecular dynamics with electronic friction at metal surfaces with numeric atom-centred orbitals", IOP Electronic Structure 5, 035005 (2023)
Design of nanostructured interfaces and thin films for single-atom catalysis and optoelectronic applications
Collaborations:
- Prof. Julie V. MacPherson, University of Warwick
- Prof. Pat Unwin, University of Warwick
- Dr. David Duncan, Diamond Light Source
- Prof. Giovanni Costantini, University of Birmingham
- Prof. Michael Gottfried, University of Marburg, Germany
Selected Publications:
- B. P. Klein, S. E. Harman, L. Ruppenthal, G. M. Ruehl, S. J. Hall, S. J. Carey, J. Herritsch, M. Schmid, R. J. Maurer, R. Tonner, C. T. Campbell, J. Michael Gottfried, “Enhanced Bonding of Pentagon–Heptagon Defects in Graphene to Metal Surfaces: Insights from the Adsorption of Azulene and Naphthalene to Pt(111)”, Chem. Mater. 32, 1041-1053 (2020).
- B. P. Klein, S. J. Hall, R. J. Maurer, "The nuts and bolts of core-hole constrained ab initio simulation for K-shell x-ray photoemission and absorption spectra", J. Phys.: Condens. Matter 33, 154005 (2021)
- D.-Q. Liu et al., "Adiabatic versus non-adiabatic electron transfer at 2D electrode materials", Nature Commun. 12, 7110 (2021)
- Knol et al., "The stabilization potential of a standing molecule", Science Advances 7, eabj9751 (2021)
- P. J. Blowey, B. Sohail, et al., "Alkali doping leads to charge-transfer salt formation in a two-dimensional metal-organic framework", ACS Nano 14, 7475-7483 (2020)