
Publications
Jump to a year:2024|2023|2022 | 2021 |2020 | 2019 |2018 | 2017 | 2016 |2015 | 2014| 2013|2012 | 2011
|
Current Funding: |
|
|
|
Scientific profiles: |
|
||||||
| Previous Funding: |
|
|
|
|
|
 co-lead
co-lead


2026
Jump to top
64. |

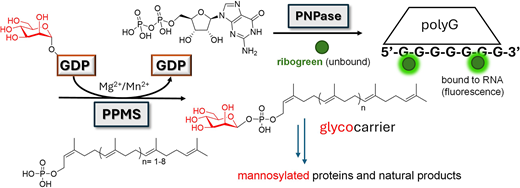
Glycosyl transfer constitutes a key step in the functionalisation of bioactive molecules, including proteins and natural products. In vitro studies to dissect glycosyltransferase catalysis are often challenging due to enzyme poor solubility, instability, and the requirement of advanced analytical set ups. Herein we report the development and use of a polynucleotide phosphorylase (PNPase)- coupled assay for the real-time monitoring of mannosyl transfer to polyprenyl phosphate carriers. This, together with molecular modeling, dynamics and site-directed mutagenesis, has allowed us to gather in depth insights into enzyme catalysis associated to health and disease.
63. |
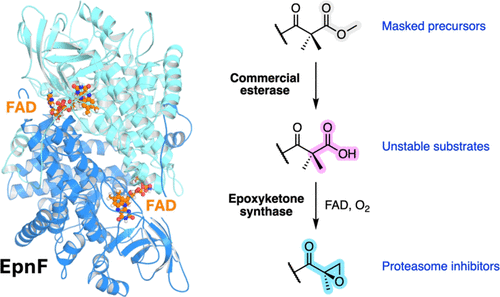
α, β-Epoxyketones are an important class of bacterial natural products with wide-ranging potential applications in oncology, immunology, and infectious disease. Their clinical potential derives from the α, β-epoxyketone pharmacophore, which covalently modifies the N-terminal catalytic threonine residue of proteasome β-subunits with high selectivity. Although a synthetic α, β-epoxyketone (Carfilzomib) is approved for clinical use, stereo-controlled synthesis of the pharmacophore remains challenging, involving either multiple steps and energy-intensive processes, or unsustainable reagents. Unusual flavoenzymes catalyze the assembly of the pharmacophore in α, β-epoxyketone biosynthesis. A detailed understanding of the substrate scope and catalytic mechanism of these enzymes has thus far been limited by the intrinsic instability of their α- (di)methyl-β-ketoacid substrates. Here, we report the development and application of an esterase-mediated unmasking strategy for in-situ generation of these substrates from the corresponding methyl esters. Using this approach, we demonstrate that EpnF, the epoxyketone synthase involved in eponemycin / TMC-86A biosynthesis, tolerates a broad range of synthetic substrate analogs, including several with N-terminal protecting groups widely used in peptide synthesis. These findings establish that EpnF has the potential to be developed into a useful biocatalyst for the chemoenzymatic synthesis of dipeptidyl epoxyketone precursors of clinically approved drugs and drug candidates. To elucidate the molecular basis for catalysis of α, β-epoxyketone formation by EpnF, substrate docking and molecular dynamics simulations were performed on a well-validated AlphaFold model, providing support for a previously proposed decarboxylation-dehydrogenation-monooxygenation mechanism. Site-directed mutagenesis and LC–MS analysis validated the proposed roles of key active-site residues in substrate positioning and catalysis of epoxide formation. Collectively, these results demonstrate that EpnF and related enzymes belong to a new class of internal flavoprotein monooxygenases and provide a foundation for developing epoxyketone synthases into useful biocatalysts for the sustainable synthesis of high-value α,β-epoxyketones.
62. |
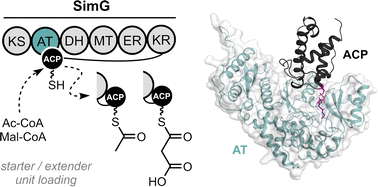
Iterative polyketide synthases (iPKSs) rely on communication between acyl carrier protein (ACP) and acyltransferase (AT) domains to ensure efficient delivery of starter and extender substrates during biosynthesis. However, the molecular determinants governing the AT:ACP interface remain poorly understood. Here, we use the fungal highly reducing PKS, SimG, a component of the cyclosporin biosynthetic pathway, as a model system to dissect the AT:ACP interface. Using alanine scanning mutagenesis combined with a high-throughput intact-protein mass spectrometry assay, we identified epitope-forming residues that affect AT:ACP interaction. These experimental constraints were used to guide docking and molecular dynamics simulations to produce a data-driven structural model of the SimG AT:ACP complex in a catalytically competent geometry. We also demonstrate that the SimG AT domain transacylates ACP domains from a range of fungal PKS architectural classes, highlighting significant interface plasticity. These insights advance our fundamental understanding of domain communication in these enigmatic megasynthases and provide a foundation for rational engineering to expand substrate scope towards novel polyketide scaffolds.
Jump to top
61. |

Polyketides and nonribosomal peptides are important natural product classes with wide-ranging medical and agricultural applications. The analogous enzymatic logic employed by bacterial modular polyketide synthases (PKSs) and nonribosomal peptide synthetases (NRPSs) enables the assembly of hybrid products. One important group of polyketide-nonribosomal peptide hybrids is exemplified by the HDAC-targeting drug romidepsin. This group is assembled by combinatorial biosynthesis involving fusion of a conserved Zn2+-binding pharmacophore to a variable peptide-based cap. Here, we use gene proximity searching to identify the FR-901375 biosynthetic gene cluster in Pseudomonas chlororaphis subsp. pisciumDSM 21509. Comparison of the PKS-NRPS encoded by this gene cluster with those assembling related depsipeptide HDAC inhibitors suggests a novel subunit docking modality enables interaction between the conserved pharmacophore and variable cap biosynthetic machineries. This hypothesis was validated using crosstalk assays, mutagenesis, AlphaFold predictions, and carbene footprinting, providing new insight into the evolution of mechanisms for hybrid polyketide-nonribosomal peptide combinatorial biosynthesis.
2025
60.
|
Glycopeptide antibiotics (GPAs) are a vital class of nonribosomal peptides used as therapies of last resort to treat infections by multidrug-resistant bacteria. These peptide antibiotics are assembled by nonribosomal peptide synthetases (NRPSs), modular megasynthases central to the biosynthesis of a wide range of peptide natural products. The adenylation (A) domains of NRPSs are involved in the selection and activation of the amino acid building blocks forming these peptide natural products, with their subsequent loading onto a neighboring carrier protein for incorporation into the growing peptide chain. This makes A-domains the gatekeepers of specificity in nonribosomal peptide biosynthesis, with further studies needed to reveal how this specificity is enforced at all stages of catalysis. The first building block found in GPAs is diverse and can comprise an amino acid, a ketoacid, or mixtures of both, which suggests that the A-domains responsible for selecting these residues can also incorporate non-amino acid substrates. In this study, we explored the acceptance of such substrates by the initiation module of the teicoplanin NRPS. Our in vitro assays demonstrated that this A-domain possesses an unexpected preference for activating ketoacids over the native amino acid substrate l-Hpg. However, only (d/l)-Hpg and related amino acids were able to be loaded onto the neighboring carrier protein domain during the subsequent thioesterification step. We further characterized the structure of this A-domain from teicoplanin biosynthesis in complex with d-4-hydroxyphenylglycine (d-Hpg), which revealed alterations in the positioning of the substrate carboxylate that help explain the high levels of pyrophosphate release seen with this amino acid. In combination with extensive molecular dynamics simulations, these data suggest that ketoacid incorporation in GPA biosynthesis is likely performed after amino acid incorporation by the NRPS and highlight the importance of considering both activation and carrier protein loading reactions performed by an A-domain when investigating substrate selectivity in nonribosomal peptide biosynthesis.
59.
Cover article
|
|---|


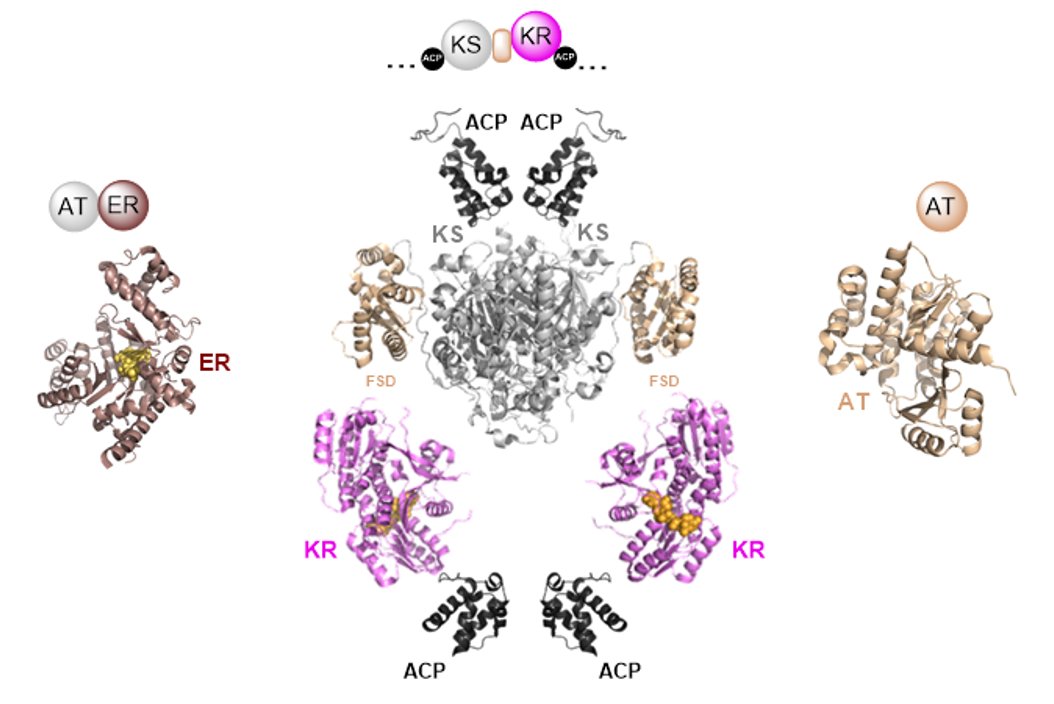
Polyketide synthases (PKSs) are multi-domain enzymatic assembly lines that biosynthesise a wide selection of bioac- tive natural products from simple building blocks. In contrast to their cis-acyltransferase (AT) counterparts, trans-AT PKSs rely on stand-alone ATs to load extender units onto acyl carrier protein (ACP) domains embedded in the core PKS machinery. Trans-AT PKS gene clusters also encode stand-alone acyl hydrolases (AHs), which are predicted to share the overall fold of ATs but function like type II thioesterases (TEIIs), hydrolysing aberrant acyl chains from ACP domains to promote biosynthetic efficiency. How AHs specifically target short acyl chains, in particular acetyl groups, tethered as thioesters to the substrate-shuttling ACP domains, with hydrolytic rather than acyl transfer activity, has remained unclear. To answer these questions, we solved the first structure of an AH and performed structure-guided activity assays on active site variants. Our results offer key insights into chain length control and selection against coenzyme A-tethered substrates, and clarify how the interaction interface between AHs and ACP domains contributes to recognition of cognate and non-cognate ACP domains. Combining our experimental findings with molecular dynamics simulations allowed for the construction of a data-driven model of an AH:ACP domain complex. Our results advance the currently incomplete understanding of polyketide biosynthesis by trans-AT PKSs, and provide foundations for future bioengineering efforts to offload biosynthetic intermediates or enhance product yields.
2024
Jump to top
58.
|
|---|

The complex multistep activation cascade of Ire1 involves changes in the Ire1 conformation and oligomeric state. Ire1 activation enhances ER folding capacity, in part by overexpressing the ER Hsp70 molecular chaperone BiP; in turn, BiP provides tight negative control of Ire1 activation. This study demonstrates that BiP regulates Ire1 activation through a direct interaction with Ire1 oligomers. Particularly, we demonstrated that the binding of Ire1 luminal domain (LD) to unfolded protein substrates not only trigger conformational changes in Ire1-LD that favour the formation of Ire1-LD oligomers but also exposes BiP binding motifs, enabling the molecular chaperone BiP to directly bind to Ire1-LD in an ATP-dependent manner. These transient interactions between BiP and two short motifs in the disordered region of Ire1-LD are reminiscent of interactions between clathrin and another Hsp70, cytoplasmic Hsc70. BiP binding to substrate-bound Ire1-LD oligomers enables unfolded protein substrates and BiP to synergistically and dynamically control Ire1-LD oligomerisation, helping to return Ire1 to its deactivated state when an ER stress response is no longer required.
2023
57.
|
|---|
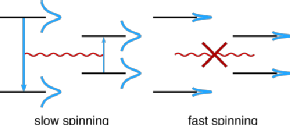
Solid-state nuclear spin diffusion is the coherent and reversible process through which spin order is transferred via dipolar couplings. With the recent increases in magic-angle spinning (MAS) frequencies and magnetic fields becoming routinely applied in solid-state nuclear magnetic resonance, understanding how the increased 1H resolution obtained affects spin diffusion is necessary for interpretation of several common experiments. To investigate the coherent contributions to spin diffusion with fast MAS, we have developed a low-order correlation in Liouville space model based on the work of Dumez et al. (J. Chem. Phys. 33, 224501, 2010). Specifically, we introduce a new method for basis set selection, which accounts for the resonance-offset dependence at fast MAS. Furthermore, we consider the necessity of including chemical shift, both isotropic and anisotropic, in the modeling of spin diffusion. Using this model, we explore how different experimental factors change the nature of spin diffusion. Then, we show case studies to exemplify the issues that arise in using spin diffusion techniques at fast spinning. We show that the efficiency of polarization transfer via spin diffusion occurring within a deuterated and 100% back-exchanged protein sample at 60 kHz MAS is almost entirely dependent on resonance offset. We additionally identify temperature-dependent magnetization transfer in beta-aspartyl L-alanine, which could be explained by the influence of an incoherent relaxation-based nuclear Overhauser effect.
2022
Jump to top
56.
|
|---|

Spin-lattice relaxation rate (R1) measurements are commonly used to characterize protein dynamics. However, the time needed to collect the data can be quite long due to long relaxation times of the low-gamma nuclei, especially in the solid state. We present a method to collect backbone heavy atom relaxation data by nesting the collection of datasets in the solid state. This method results in a factor of 2 to 2.5 times faster data acquisition for backbone R1 relaxation data for the 13C and 15N sites of proteins.
55. |
|---|
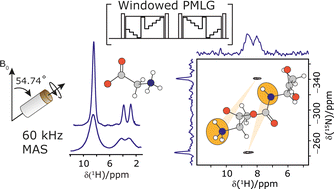
The Lee–Goldburg condition for homonuclear decoupling in 1H magic-angle spinning (MAS) solid-state NMR sets the angle θ, corresponding to arctan of the ratio of the rf nutation frequency, ν1, to the rf offset, to be the magic angle, θm, equal to tan−1(√2) = 54.7°. At 60 kHz MAS, we report enhanced decoupling compared to MAS alone in a 1H spectrum of 15N-glycine with 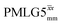 at θ = 30° for a ν1 of ∼100 kHz at a 1H Larmor frequency, ν0, of 500 MHz and 1 GHz, corresponding to a high chemical shift scaling factor (λCS) of 0.82. At 1 GHz, we also demonstrate enhanced decoupling compared to 60 kHz MAS alone for a lower ν1 of 51 kHz, i.e., a case where the nutation frequency is less than the MAS frequency, with θ = 18°, λCS = 0.92. The ratio of the rotor period to the decoupling cycle time, Ψ = τr/τc, is in the range 0.53 to 0.61. Windowed
at θ = 30° for a ν1 of ∼100 kHz at a 1H Larmor frequency, ν0, of 500 MHz and 1 GHz, corresponding to a high chemical shift scaling factor (λCS) of 0.82. At 1 GHz, we also demonstrate enhanced decoupling compared to 60 kHz MAS alone for a lower ν1 of 51 kHz, i.e., a case where the nutation frequency is less than the MAS frequency, with θ = 18°, λCS = 0.92. The ratio of the rotor period to the decoupling cycle time, Ψ = τr/τc, is in the range 0.53 to 0.61. Windowed 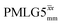 decoupling using the optimised parameters for a ν1 of ∼100 kHz also gives good performance in a 1H spin-echo experiment, enabling implementation in a 1H-detected 15N–1H cross polarisation (CP)-refocused INEPT heteronuclear correlation NMR experiment. Specifically, initial 15N transverse magnetisation as generated by 1H–15N CP is transferred back to 1H using a refocused INEPT pulse sequence employing windowed
decoupling using the optimised parameters for a ν1 of ∼100 kHz also gives good performance in a 1H spin-echo experiment, enabling implementation in a 1H-detected 15N–1H cross polarisation (CP)-refocused INEPT heteronuclear correlation NMR experiment. Specifically, initial 15N transverse magnetisation as generated by 1H–15N CP is transferred back to 1H using a refocused INEPT pulse sequence employing windowed 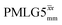 1H decoupling. Such an approach ensures the observation of through-bond N–H connectivities. For 15N-glycine, while the CP-refocused INEPT experiment has a lower sensitivity (∼50%) as compared to a double CP experiment (with a 200 μs 15N to 1H CP contact time), there is selectivity for the directly bonded NH3+ moiety, while intensity is observed for the CH21H resonances in the double CP experiment. Two-dimensional 15N–1H correlation MAS NMR spectra are presented for the dipeptide β-AspAla and the pharmaceutical cimetidine at 60 kHz MAS, both at natural isotopic abundance. For the dipeptide β-AspAla, different build-up dependence on the first spin-echo duration is observed for the NH and NH3+ moieties demonstrating that the experiment could be used to distinguish resonances for different NHx groups.
1H decoupling. Such an approach ensures the observation of through-bond N–H connectivities. For 15N-glycine, while the CP-refocused INEPT experiment has a lower sensitivity (∼50%) as compared to a double CP experiment (with a 200 μs 15N to 1H CP contact time), there is selectivity for the directly bonded NH3+ moiety, while intensity is observed for the CH21H resonances in the double CP experiment. Two-dimensional 15N–1H correlation MAS NMR spectra are presented for the dipeptide β-AspAla and the pharmaceutical cimetidine at 60 kHz MAS, both at natural isotopic abundance. For the dipeptide β-AspAla, different build-up dependence on the first spin-echo duration is observed for the NH and NH3+ moieties demonstrating that the experiment could be used to distinguish resonances for different NHx groups.
2021
Jump to top
54. |
|---|
Order parameters are a useful tool for quantifying amplitudes of molecular motions. Here we measure dipolar order parameters by recoupling heteronuclear dipole-dipole couplings under fast spinning. We apply symmetry based recoupling methods to samples spinning under magic angle at 60 kHz by employing a variable flip angle compound inversion pulse. We validate the methods by measuring site-specific 15N-1H order parameters of a microcrystalline protein over a small temperature range and the same protein in a large, precipitated complex with antibody. The measurements of the order parameters in the complex are consistent with the observed protein undergoing overall motion within the assembly.
53.
Featured on the inside front cover |
Munro Passmore; Angelo Gallo; Józef R. Lewandowski; Matthew Jenner "Molecular Basis for Acyl Carrier Protein-Ketoreductase Interaction in trans-Acyltransferase Polyketide Synthases" Chemical Science 2021,12, 13676-13685. DOI: https://doi.org/10.1039/D1SC03478B (accepted 29 Sept 2021, online ) | WRAP: Data (200 ns classical MD simulations of PksJ ACP4:KR2 complex): http://dx.doi.org/10.17632/396hfrrpkw.1 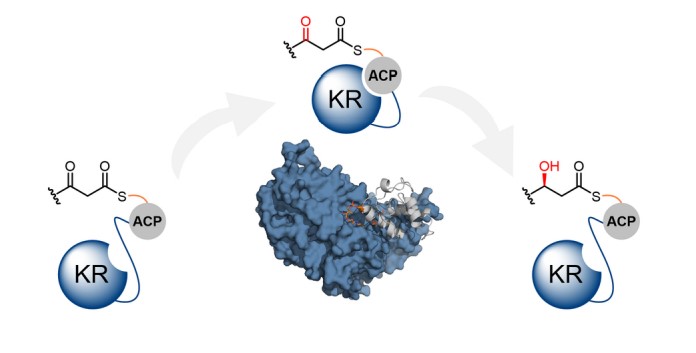 Keywords: natural products biosynthesis |
|---|

The biosynthesis of polyketides by type I modular polyketide synthases (PKSs) relies on co-ordinated interactions between acyl carrier protein (ACP) domains and catalytic domains within the megasynthase. Despite the importance of these interactions, and their implications for biosynthetic engineering efforts, they remain poorly understood. Here, we report the molecular details of the interaction interface between an ACP domain and a ketoreductase (KR) domain from a transacyltransferase (trans-AT) PKS. Using a high-throughput mass spectrometry (MS)-based assay in combination with scanning alanine mutagenesis, residues contributing to the KR-binding epitope of the ACP domain were identified. Application of carbene footprinting revealed the ACP-binding site on the KR domain surface, and molecular docking simulations driven by experimental data allowed production of an accurate model of the complex. Interactions between ACP and KR domains from trans-AT PKSs were found to be specific for their cognate partner, indicating highly optimised interaction interfaces driven
by evolutionary processes. Using detailed knowledge of the ACP:KR interaction epitope, an ACP domain was engineered to interact with a non-cognate KR domain partner. The results provide a novel, high resolution insights into the ACP:KR interface and offer valuable rules for future engineering efforts of biosynthetic assembly lines.
52.
Featured on the cover. |
Patrick M.J. Szell, Józef R. Lewandowski, Helen Blade, Leslie P. Hughes, Sten O. Nilsson Lill, Steven P. Brown. "Taming the dynamics in a pharmaceutical by cocrystallization: investigating the impact of the coformer by solid-state NMR" CrystEngComm 2021. DOI: https://doi.org/10.1039/D1CE01084K (accepted 24 Aug 2021, online 2 Sept 2021 ) | WRAP: Data on WRAP: http://wrap.warwick.ac.uk/157143/ 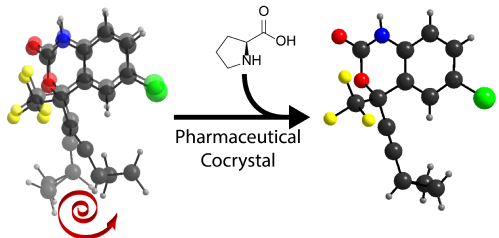 Keywords: solid-state NMR; pharmaceuticals; dynamics |
|---|

The motion of functional groups occurring in pharmaceuticals, known as dynamics, can affect their chemical stability and physicochemical properties, while complicating the process of structural characterization. Here, we show the first instance of controlling the dynamics of a pharmaceutical via cocrystallization, using the anti-HIV drug Efavirenz (1) as a model. While the crystalline form of 1 exhibits motion in its cyclopropyl and trifluoromethyl groups, with further secondary motion across the entire molecule, the cocrystals are much less dynamic. In the case of the cocrystal with L-proline, sample 1e, the structure appears to be rigid. This effect originates from favourable crystal packing induced by the coformer, constraining the cyclopropyl ring in place. With pharmaceutical cocrystals continuing to gain interest both academically and commercially, these insights may allow the physicochemical properties of pharmaceutical cocrystal to be understood.
51.
|
Tognetti, Jacqueline; Franks, W. Trent; Gallo, Angelo; Lewandowski, Józef R. "Accelerating 15N and 13C R1 and R1ρ relaxation measurements by multiple pathway solid-state NMR experiments " J Magn Reson 2021, 331, 107049. DOI: https://doi.org/10.1016/j.jmr.2021.107049Link opens in a new window (accepted 11 Aug 2021, online ) | WRAP: Pulse sequences and raw data: 10.17632/x7kk4rkpj3.1 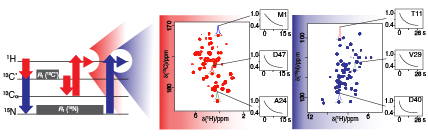
Keywords: relaxation; solid-state NMR; accelerated methods |
|---|

Magic angle spinning (MAS) Solid-state NMR is a powerful technique to probe dynamics of biological systems at atomic resolution. R1 and R1ρ relaxation measurements can provide detailed insight on amplitudes and time scales of motions, especially when information from several different site-specific types of probes is combined. However, such experiments are time-consuming to perform. Shortening the time necessary to record relaxation data for different nuclei will greatly enhance practicality of such approaches. Here, we present staggered acquisition experiments to acquire multiple relaxation experiments from a single excitation to reduce the overall experimental time. Our strategy enables one to collect 15N and 13C relaxation data in a single experiment in a fraction of the time necessary for two separate experiments, with the same signal to noise ratio.
50.
|
Fage, Christopher; Kosol, Simone; Jenner, Matthew; Öster, Carl; Gallo, Angelo; Kaniusaite, Milda; Steinbach, Roman; Staniforth, Michael; Stavros, Vasilios; Marahiel, Mohamed; Cryle, Max; Lewandowski, Józef R. "Communication Breakdown: Dissecting the COM Interfaces between the Subunits of Nonribosomal Peptide Synthetases" ACS Catalysis 2021, 11, 17, 10802–10813. DOI: https://doi.org/10.1021/acscatal.1c02113 (accepted 27 July 2021, online ) | WRAP: http://wrap.warwick.ac.uk/157197/ Abridged MD trajectories:
Keywords: natural products; NRPS; communication domains; docking domains; folding; intrinsically disordered regions; solution NMR; solid-state NMR; X-ray crystallography; carbene footprinting; molecular dynamics simulations; biochemistry; molecular biology; bioinformatics; |
|---|

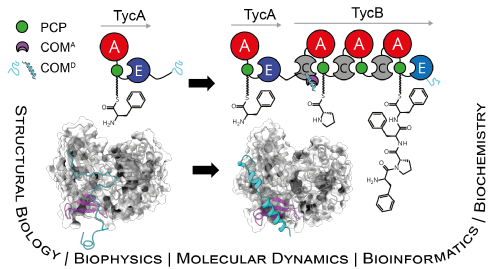
Nonribosomal peptides are a structurally diverse and bioactive class of natural products constructed by multidomain enzymatic assembly lines known as nonribosomal peptide synthetases (NRPSs). While the core catalytic domains and even entire protein subunits of NRPSs have been structurally elucidated, little biophysical work has been reported on the docking domains that promote interactions—and thus transfer of biosynthetic intermediates—between subunits. In the present study, we closely examine the COM domains that mediate COMmunication between donor epimerization (E) and acceptor condensation (C) domains found at the termini of NRPS subunits. Through a combination of X-ray crystallography, circular dichroism spectroscopy, solution- and solid-state NMR spectroscopy, and molecular dynamics (MD) simulations, we provide the first direct evidence for an intrinsically disordered donor COM region that folds into a dynamic helical motif upon binding to a suitable acceptor. Furthermore, our NMR titration and carbene footprinting experiments illuminate the residues involved at the COM interaction interface, and our MD simulations demonstrate folding consistent with experimental data. Although our results lend credence to the previously proposed helix-hand mode of interaction, they also underscore the importance of viewing COM interfaces as dynamic ensembles rather than single rigid structures and suggest that engineering experiments should account for the interactions which transiently guide folding in addition to those which stabilize the final complex. Through activity assays and affinity measurements, we further substantiate the role of the donor COM region in binding the acceptor C domain and implicate this short motif as readily transposable for non-cognate domain crosstalk. Finally, our bioinformatics analyses show that COM domains are widespread in natural product pathways and function at interfaces beyond the canonical type described above, setting a high priority for thorough characterization of these docking domains. Our findings lay the groundwork for future attempts to rationally engineer new NRPS domain-domain interactions with the ultimate goal of generating new bioactive molecules.
49.
|
|---|

Polyketide synthase (PKS) and non-ribosomal peptide synthetase (NRPS) multienzymes produce numerous high value metabolites. The protein subunits which constitute these megasynth(et)ases must undergo ordered self-assembly to ensure correct organisation of catalytic domains for the biosynthesis of a given natural product. Short amino acid regions at the N- and C-terminus of each subunit, termed docking domains (DDs), often occur in complementary pairs, which interact to facilitate substrate transfer and maintain pathway fidelity. This review details all structurally characterised examples of NRPS and PKS DDs to date and summarises efforts to utilise DDs for the engineering of biosynthetic pathways.
2020
Jump to top
48.
The manuscript featured on the journal cover. |
Samuel J. Page, Angelo Gallo, Steven P. Brown, Józef R. Lewandowski, John V. Hanna, W. Trent Franks "Simultaneous MQMAS NMR Experiments for Two Half-Integer Quadrupolar Nuclei" J. Magn. Reson. 2020, 320, 106831. DOI: https://doi.org/10.1016/j.jmr.2020.106831Link opens in a new window (accept. 18.09.20, online 23.09.20) | WRAP: 
Keywords: methodology; multiple receivers; quadrupole nuclei; MQMAS; solid-state NMR |
|---|

A procedure to acquire two Multiple-Quantum Magic Angle Spinning (MQMAS) NMR experiments with the same instrument time is presented. A triply tuned probe is utilized with multiple receivers to collect data with staggered acquisitions and thus more efficiently use the instrument time. The data for one nucleus is collected during the relaxation delay of the other nucleus, and vice versa. The instrument time is reduced to 60-80% of the time needed for the single acquisition collection Specifically our approach is presented for recording triple quantum (3Q) 17O and either 3Q or quintuple quantum (5Q) 27Al NMR spectra of a 1.18Na2O•5SiO2•Al2O3 glass gel.
47.
|
Alexander N. Baker, Sarah-Jane Richards, Collette S. Guy, Thomas R. Congdon, Muhammad Hasan, Alexander J. Zwetsloot, Angelo Gallo, Jozef R. Lewandowski, Phillip J. Stansfeld, Anne Straube, Marc Walker, Simona Chessa, Giulia Pergolizzi, Simone Dedola, Robert A. Field, and Matthew I. Gibson "The SARS-COV‑2 Spike Protein Binds Sialic Acids and Enables Rapid DOI: https://doi.org/10.1021/acscentsci.0c00855 | WRAP: Beautiful work from Gibson group with small contribution from our group.
Keywords: applications; STD; solution NMR |
|---|


There is an urgent need to understand the behavior of the novel coronavirus (SARS-COV-2), which is the causative agent of COVID-19, and to develop point-of-care diagnostics. Here, a glyconanoparticle platform is used to discover that N-acetyl neuraminic acid has affinity toward the SARS-COV-2 spike glycoprotein, demonstrating its glycan-binding function. Optimization of the particle size and coating enabled detection of the spike glycoprotein in lateral flow and showed selectivity over the SARS-COV-1 spike protein. Using a virus-like particle and a pseudotyped lentivirus model, paper-based lateral flow detection was demonstrated in under 30 min, showing the potential of this system as a low-cost detection platform.
46.
Cover art designed by Sarah Mann |
Sarah K. Mann, Mohit K. Devgan, W. Trent Franks, Steven Huband, Chi Long Chan, Jeraime Griffith, David Pugh, Nicholas J. Brooks, Tom Welton, Tran N. Pham, Lisa L. McQueen, Józef R. Lewandowski*, and Steven P. Brown* "MAS NMR Investigation of Molecular Order in an Ionic Liquid Crystal" J. Phys. Chem. B 2020 124, 24, 4975–4988. DOI: https://doi.org/10.1021/acs.jpcb.0c02328 | WRAP: Collaboration with Brown and Welton groups and GSK.
Keywords: solid-state NMR; solution NMR; methodology; ionic liquids |
|---|



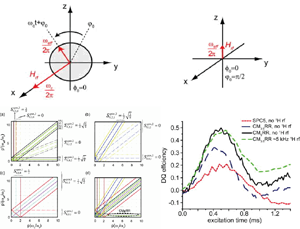
The structure and molecular order in the thermotropic ionic liquid crystal (ILC), [choline][geranate(H)octanoate], an analogue of Choline And GEranate (CAGE), which has potential for use as a broad-spectrum antimicrobial and transdermal and oral delivery agent, were investigated by magic-angle spinning (MAS) nuclear magnetic resonance (NMR), polarizing optical microscopy, small-angle X-ray scattering (SAXS), and mass spectrometry. Mass spectrometry and the 1H NMR chemical shift reveal that CAGE-oct is a dynamic system, with metathesis (the exchange of interacting ions) and hydrogen exchange occurring between hydrogen-bonded/ionic complexes such as [(choline)(geranate)(H)(octanoate)], [(choline)(octanoate)2(H)], and [(choline)(geranate)2(H)]. These clusters, which are shown by mass spectrometry to be significantly more stable than expected for typical electrostatic ion clusters, involve hydrogen bonding between the carboxylic acid, carboxylate, and hydroxyl groups, with rapid hydrogen bond breaking and re-formation observed to average the 1H chemical shifts. The formation of a partial bilayer liquid crystal (LC) phase was identified by SAXS and polarizing optical microscopy at temperatures below ∼293 K. The occurrence of this transition close to room temperature could be utilized as a potential temperature-induced “switch” of the anisotropic properties for particular applications. The presence of an isotropic component of approximately 23% was observed to coexist with the LC phase, as detected by polarizing optical microscopy and quantified by both 1H–13C dipolar-chemical shift correlation (DIPSHIFT) and 1H double-quantum (DQ) MAS NMR experiments. At temperatures above the LC-to-isotropic transition, intermediate-range order (clustering of polar and nonpolar domains), a feature of many ILs, persists. Site-specific order parameters for the LC phase of CAGE-oct were obtained from the MAS NMR measurement of the partially averaged 13C–1H dipolar couplings (DCH) by cross-polarization (CP) build-up curves and DIPSHIFT experiments, and 1H–1H dipolar couplings (DHH) by double-quantum (DQ) build-up curves. The corresponding order parameters, SCH and SHH, are in the range 0–0.2 and are lower compared to those for smectic (i.e., layered) phases of conventional nonionic liquid crystals, resembling those of lamellar phases formed by lyotropic surfactant–solvent systems.
45. |
Matthew R. Gyton, Amy E. Kynman, Baptiste Leforestier, Angelo Gallo, Józef R. Lewandowski and Adrian B. Chaplin "Isolation and structural characterisation ofrhodium(III)η2-fluoroarene complexes: experi-mental verification of predicted regioselectivity" Dalton Transactions 2020 49, 5791. DOI: 10.1039/d0dt01137a | WRAP: Collaboration with Chaplin group. 
Keywords: solid-state NMR; applications; fluorine |
|---|

The isolation and solid-state characterisation of complexes featuring partially coordinated benzene, fluorobenzene and all three isomers of difluorobenzene are described. Supported by a DFT analysis, this well-defined homologous series demonstrates the preference for η2-coordination of fluoroarenes via the HCvCH sites adjacent to a fluorine substituent.
44.
|
|---|

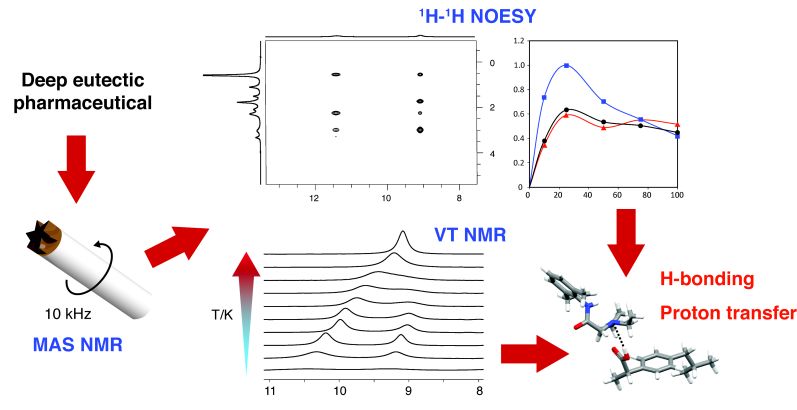
Liquid forms of pharmaceuticals (ionic liquids and deep eutectic solvents) offer a number of potential advantages over solid-state drugs; a key question is the role of intermolecular hydrogen bonding interactions in enabling membrane transport. Characterisation is challenging since high sample viscosities, typical of liquid pharmaceutical formulations, hamper the use of conventional solution NMR at ambient temperature. Here, we report the application of magic-angle spinning (MAS) NMR spectroscopy to the deep eutectic pharmaceutical, lidocaine ibuprofen. Using variable temperature MAS NMR, the neat system, at a fixed molar ratio, can be studied over a wide range of temperatures, characterized by changing mobility, using a single experimental setup. Specific intermolecular hydrogen bonding interactions are identified by two-dimensional 1H-1H NOESY and ROESY MAS NMR experiments. Hydrogen-bonding dynamics are quantitatively determined by following the chemical exchange process between the labile protons by means of line-width analysis of variable temperature 1H MAS NMR spectra.
2019
Jump to top
43.
|
|---|

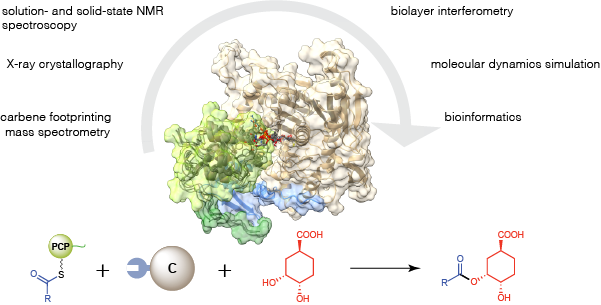
Modular polyketide synthases and non-ribosomal peptide synthetases are molecular assembly lines that consist of several multienzyme subunits that undergo dynamic self-assembly to form a functional megacomplex. N- and C-terminal docking domains are usually responsible for mediating the interactions between subunits. Here we show that communication between two non-ribosomal peptide synthetase subunits responsible for chain release from the enacyloxin polyketide synthase, which assembles an antibiotic with promising activity against Acinetobacter baumannii, is mediated by an intrinsically disordered short linear motif and a β-hairpin docking domain. The structures, interactions and dynamics of these subunits were characterized using several complementary biophysical techniques to provide extensive insights into binding and catalysis. Bioinformatics analyses reveal that short linear motif/β-hairpin docking domain pairs mediate subunit interactions in numerous non-ribosomal peptide and hybrid polyketide–non-ribosomal peptide synthetases, including those responsible for assembling several important drugs. Short linear motifs and β-hairpin docking domains from heterologous systems are shown to interact productively, highlighting the potential of such interfaces as tools for biosynthetic engineering.
42.
|
|---|

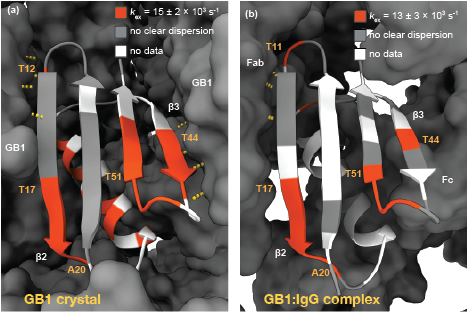
Solid state NMR is a powerful method to obtain information on the structure and dynamics of protein complexes that, due to solubility and size limitations, cannot be achieved by other methods. Here, we present an approach that allows the quantification of microsecond conformational exchange in large protein complexes by using a paramagnetic agent to accelerate 15N R1ρ relaxation dispersion measurements and overcome sensitivity limitations. The method is validated on crystalline GB1 and then applied to a > 300 kDa precipitated complex of GB1 with full length human immunoglobulin G (IgG). The addition of a paramagnetic agent increased the signal to noise ratio per time unit by a factor of 5, which allowed full relaxation dispersion curves to be recorded on a sample containing less than 50 μg of labelled material in 5 and 10 days on 850 and 700 MHz spectrometers, respectively. We discover a similar exchange process across the β-sheet in GB1 in crystals and in complex with IgG. However, the slow motion observed for a number of residues in the α-helix of crystalline GB1 is not detected in the complex.
41.
|


We present a suite of two-receiver solid-state NMR experiments for backbone and side chain resonance assignment. The experiments rely on either dipolar coupling or scalar coupling for polarization transfer and are devised to acquire a 1H–detected 3D experiment AND a nested 13C–detected 2D from a shared excitation pulse. In order to compensate for the lower sensitivity of detection on 13C nucleus, 2D rows are signal averaged during 3D planes. The 3D dual receiver experiments do not suffer from any appreciable signal loss compared to their single receiver versions and require no extra optimization. The resulting data is higher in information content with no additional experiment time. The approach is expected to become widespread as multiple receivers become standard for new NMR spectrometers.
40.
|

![]()
The activation mechanism of the ErbB family of receptors is of considerable medical interest as they are linked to a number of human cancers, including an aggressive form of breast cancer. In the rat analogue of the human ErbB2 receptor, referred to as Neu, a point mutation in the transmembrane domain (V664E) has been shown to trigger oncogenic transformation. While the structural impact of this mutation has been widely studied in the past to yield models for the active state of the Neu receptor, little is known about the impact of cholesterol on its structure. Given previous reports of the influence of cholesterol on other receptor tyrosine kinases (RTKs), as well as the modulation of lipid composition in cancer cells, we wished to investigate how cholesterol content impacts the structure of the Neu transmembrane domain. We utilized high-resolution magic angle spinning solid-state NMR to measure 13C–13C coupling of selectively labelled probe residues in the Neu transmembrane domain in lipid bilayers containing cholesterol. We observe inter-helical coupling between residues that support helix–helix interactions on both dimerization motifs reported in the literature (A661-XXX-G665 and I659-XXX-V663). We further explore how changes in cholesterol concentration alter transmembrane domain interactions and the properties and mechanics of the bilayer. We interpret our results in light of previous studies relating RTK activity to cholesterol enrichment and/or depletion, and propose a novel model to explain our data that includes the recognition and binding of cholesterol by the Neu transmembrane domain through a putative cholesterol-recognition/interaction amino acid consensus sequence.
2018
Jump to top
39.
|
L.M. Alkhalaf, S.M. Barry, D. Ray, A. Gallo, D. Griffths, J. R. Lewandowski, V. Fulop and G.L. Challis. "Binding of distinct substrate conformations enables hydroxylation of remote sites in thaxtomin D by cytochrome P450 TxtC." J. Am. Chem. Soc. (2018) 141 (1), 216–222. DOI: 10.1021/jacs.8b08864 |

Cytochromes P450 (CYPs) catalyze various oxidative transformations in drug metabolism, xenobiotic degradation and natural product biosynthesis. Here we report biochemical, structural and theoretical studies of TxtC, an unusual bifunctional CYP in-volved in the biosynthesis of the EPA-approved herbicide thaxtomin A. TxtC was shown to hydroxylate two remote sites with-in the Phe residue of its diketopiperazine substrate thaxtomin D. The reactions follow a preferred order, with hydroxylation of the α-carbon preceding functionalization of the phenyl group. To illuminate the molecular basis for remote site functionaliza-tion, X-ray crystal structures of TxtC in complex with the substrate and monohydroxylated intermediate were determined. Electron density corresponding to a diatomic molecule (probably dioxygen) was sandwiched between the heme iron atom and Thr237 in the TxtC-intermediate structure, providing insight into the mechanism for conversion of the ferrous-dioxygen com-plex into the reactive ferryl intermediate. The substrate and monohydroxylated intermediate adopted similar conformations in the active site, with the π-face of the phenyl group positioned over the heme iron atom. Docking simulations reproduced this observation and identified a second, energetically similar but conformationally-distinct binding mode in which the α-hydrogen of the Phe residue is positioned over the heme prosthetic group. Molecular dynamics simulations confirmed that the α-hydrogen is sufficiently close to the ferryl oxygen atom to be extracted by it and indicated that the two substrate conformations cannot readily interconvert in the active site. These results indicate that TxtC is able to hydroxylate two spatially remote sites by binding distinct conformations of the substrate and monohydroxylated intermediate.
38.
|
B. Busi, J. R. Yarava, A. Hofstetter, N. Salvi, D. Cala-De Paepe, J. R. Lewandowski, M. Blackledge, and L. Emsley "Protein Dynamics from Multi-Field Variable Temperature NMR Relaxation and MD Simulation" J. Phys. Chem. B (2018) 122 (42), 9697–9702. DOI: 10.1021/acs.jpcb.8b08578 Submitted: 3 Sept 2018, Accepted: 2 Oct 2018, Published online: 2 Oct 2018 |


Understanding the interplay between protein function and dynamics is currently one of the fundamental challenges of physical biology. Recently a method using variable temperature solid state nuclear magnetic resonance relaxation measurements was proposed for the simultaneous measurement of 12 different activation energies reporting on distinct dynamic modes in the protein GB1. Here, we extend this approach to measure relaxation at multiple magnetic field strengths, allowing us to better constrain the motional models, and to simultaneously evaluate the robustness and physical basis of the method. The data reveal backbone and sidechain motions exhibiting low and high-energy modes with temperature coefficients around 5 kJ·mol-1 and 25 kJ·mol-1. The results are compared to variable temperature molecular dynamics simulation of the crystal lattice, providing further support for the interpretation of the experimental data in terms of molecular motion.
37.
|
Review S. Kosol, M. Jenner, J. R. Lewandowski, G. L. Challis "Protein–protein interactions in trans-AT polyketide synthases" Nat. Prod. Rep. (2018) 35, 1097-1109. DOI://10.1039/C8NP00066B Submitted: 26 July 2018, Published online: 3 Oct 2018 |
|

The construction of polyketide natural products by type I modular polyketide synthases (PKSs) requires the coordinated action of several protein subunits to ensure biosynthetic fidelity. This is particularly the case for trans-AT PKSs, which in contrast to most cis-AT PKSs, contain split modules and employ several transacting catalytic domains. This article summarises recent advances in understanding the protein–protein interactions underpinning subunit assembly and intra-subunit communication in such systems and highlights potential avenues and approaches for future research.
36. |
C. Öster, G. P. Walkowiak, D. E. Hughes, A. L. Spoering, A. J. Peoples, A. C. Catherwood, J. A. Tod, A. J. Lloyd, T. Herrmann, K. Lewis, C. G. Dowson, Józef R. Lewandowski "Structural studies suggest aggregation as one of the modes of action for teixobactin" Chem. Sci. (2018) 9, 8850-8859. DOI: 10.1039/C8SC03655A Submitted: 15 Jun 2018 (rev.: 16 Aug 2018), Accepted: 19 Sept 2018, Published online: 20 Sept 2018 | Open Access | Available from WRAP Associated data: Raw NMR data: http://wrap.warwick.ac.uk/108554/ | BMRB entries: 27478 (solution; teixobactin in DPC micelles), 27479 (solution; teixobactin in aqueous solution) and 27480 (teixobactin in a sedimeneted complex with Gram-negative lipid II in a presence of DPC) | Structure of teixobactin in DPC micelles PDB |


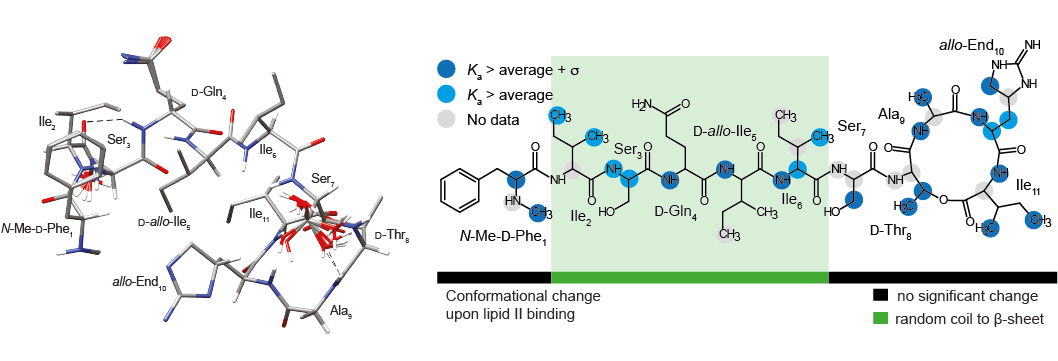
Teixobactin is a new promising antibiotic that targets cell wall biosynthesis by binding to lipid II and has no detectable resistance thanks to its unique but yet not fully understood mechanism of operation. To aid in structure-based design of teixobactin analogues with improved pharmacological properties, we present a 3D structure of native teixobactin in membrane mimetics and characterise its binding to lipid II through a combination of solution NMR and fast (90 kHz) magic angle spinning solid state NMR. In NMR titrations, we observe a pattern strongly suggesting interactions between the backbone of the C-terminal “cage” with the pyrophosphate moiety in lipid II. We find that the N-terminal part of teixobactin does not only act as a membrane anchor, as previously thought, but is actively involved in binding. Moreover, teixobactin forms a well-structured and specific complex with lipid II, where the N-terminal part of teixobactin assumes a β conformation that is highly prone to aggregation, which likely contributes to the antibiotic’s high bactericidal efficiency. Overall, our study provides several new clues to teixobactin’s modes of action.
35.
H line width dependence on MAS speed in solid state NMR - Comparison of experiment and simulation. |
U. Sternberg, R. Witter, I. Kuprov, J.M. Lamley, A. Oss, J.R. Lewandowski and A. Samoson "1H Line Width Dependence on MAS Speed in Solid State NMR - Comparison of Experiment and Simulation" J. Magn. Reson. (2018) 291, 32-39. DOI://10.1016/j.jmr.2018.04.003 (Open Access article) #1 most read JMR article of 2018 Submitted: 30 Nov 2017 (rev.: 4 Apr 2018), Accepted: 6 Apr 2018, Published online: 7 Apr 2018 | Open Access | Available from WRAP Associated data: https://data.mendeley.com/datasets/rrc9pzc4wk/1 |

Recent developments in magic angle spinning (MAS) technology permit spinning frequencies of ≥100 kHz. The effect of such fast MAS rates upon nuclear magnetic resonance (NMR) proton line widths on the multiple 1H-spin-system of β-Asp-Ala crystal structure had been examined. Powder pattern simulations were performed applying Fokker-Plank approach as implemented in Spinach with periodic boundary conditions, using crystal coordinates and 1H-chemical shift tensors calculated using the bond polarization theory (BPT). The theoretical predictions mirror well the experimental results. Both approaches demonstrate that homogeneous broadening has a linear-quadratic dependency on the inverse of the MAS spinning frequency and that, at the faster end of the spinning frequencies, the residual spectral line broadening becomes dominated by chemical shift distributions and susceptibility effects even for crystalline systems.
34.
|
M. Jenner, S. Kosol, D. Griffiths, P. Prasongpholchai, L. Manzi, A.S. Barrow, J. E. Moses, N. J. Oldham, J. R. Lewandowski, G. L. Challis "Mechanism of intersubunit ketosynthase–dehydratase interaction in polyketide synthases." Nat. Chem. Biol. (2018) 14(3), 270-275. DOI:10.1038/nchembio.2549 Submitted: 30 Nov 2017 (rev.: 4 Apr 2018), Accepted: 6 Apr 2018, Published online: 7 Apr 2018 | Accepted version freely available from WRAP |



Modular polyketide synthases (PKSs) produce numerous structurally complex natural products that have diverse applications in medicine and agriculture. PKSs typically consist of several multienzyme subunits that utilize structurally defined docking domains (DDs) at their N and C termini to ensure correct assembly into functional multiprotein complexes. Here we report a fundamentally different mechanism for subunit assembly in trans-acyltransferase (trans-AT) modular PKSs at the junction between ketosynthase (KS) and dehydratase (DH) domains. This mechanism involves direct interaction of a largely unstructured docking domain (DD) at the C terminus of the KS with the surface of the downstream DH. Acyl transfer assays and mechanism-based crosslinking established that the DD is required for the KS to communicate with the acyl carrier protein appended to the DH. Two distinct regions for binding of the DD to the DH were identified using NMR spectroscopy, carbene footprinting, and mutagenesis, providing a foundation for future elucidation of the molecular basis for interaction specificity.
Associated data: assignment for GbnD4 KS14 DHDD BMRB 27016
2017| Jump to top
33. C. Öster, S. Kosol, C. Hartlmüller, J.M. Lamley, D. Iuga, A. Oss, M.-L. Org, K. Vanatalu, A. Samoson, T. Madl, J.R. Lewandowski "Characterization of protein-protein interfaces in large complexes by solid state NMR solvent paramagnetic relaxation enhancements" J. Am. Chem. Soc. (2017) 139 (35), 12165–12174. DOI: 10.1021/jacs.7b03875 (Open Access Article) |
Raw NMR data available from: http://wrap.warwick.ac.uk/91300/1/All_data_sPRE.zip
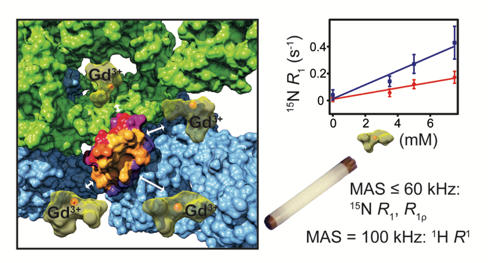
Solid-state NMR is becoming a viable alternative for obtaining information about structures and dynamics of large biomolecular complexes including ones that are not accessible to other high resolution biophysical techniques. In this context, methods for probing protein-protein interfaces at atomic resolution are highly desirable. Solvent paramagnetic relaxation enhancements (sPREs) proved to be a powerful method for probing protein-protein interfaces in large complexes in solution but have not been employed towards this goal in the solid state. We demonstrate that 1H and 15N relaxation-based sPREs provide a powerful tool for characterizing intermolecular interactions in large assemblies in the solid state. We present approaches for measuring sPREs in practically the entire range of magic angle spinning frequencies used for biomolecular studies and discuss their benefits and limitations. We validate the approach on crystalline GB1 with our experimental results in good agreement with theoretical predictions. Finally, we use sPREs to characterize protein-protein interfaces in the GB1 complex with immunoglobulin (IgG). Our results suggest the potential existence of an additional binding site and provide new insights into GB1:IgG complex structure that amend and revise the current model available from studies with IgG fragments. We demonstrate sPREs as a practical, widely applicable, robust and very sensitive technique for determining intermolecular interaction interfaces in large biomolecular complexes in the solid state.
| 32. Good, D., Pham, C., Jagas, J., Lewandowski, J.R and Ladizhansky V. "Solid-state NMR provides evidence for small-amplitude slow domain motions in a multi-spanning transmembrane α-helical protein" J. Am. Chem. Soc. (2017) 139 (27), 9246–9258. DOI: 10.1021/jacs.7b03974 (Open Access Publication) |

| Proteins are dynamic entities and populate ensembles of conformations. Transitions between states within a conformational ensemble occur over a broad spectrum of amplitude and time scales, and are often related to biological function. Whereas solid-state NMR (SSNMR) spectroscopy has recently been used to characterize conformational ensembles of proteins in the microcrystalline states, its applications to membrane proteins remain limited. Here we use SSNMR to study conformational dynamics of a seven-helical transmembrane (TM) protein, Anabaena Sensory Rhodopsin (ASR) reconstituted in lipids. We report on site-specific measurements of the 15N longitudinal R1 and rotating frame R1ρ relaxation rates at two fields of 600 and 800 MHz and at two temperatures of 7 and 30 °C. Quantitative analysis of the R1 and R1ρ values and of their field and temperature dependencies provides evidence of motions on at least two time scales. We modeled these motions as fast local motions and slower collective motions of TM helices and of structured loops, and used the simple model-free and extended model-free analyses to fit the data and estimate the amplitudes, time scales and activation energies. Faster picosecond (tens to hundreds of picoseconds) local motions occur throughout the protein and are dominant in the middle portions of the TM helices. In contrast, the amplitudes of the slower collective motions occurring on the nanosecond (tens to hundreds of nanoseconds) time scales, are smaller in the central parts of helices, but increase toward their cytoplasmic sides as well as in the interhelical loops. ASR interacts with a soluble transducer protein on its cytoplasmic surface, and its binding affinity is modulated by light. The larger amplitude of motions on the cytoplasmic side of the TM helices correlates with the ability of ASR to undergo large conformational changes in the process of binding/unbinding the transducer. |
2016| Jump to top
31. Lamley, J.M., Lewandowski, J. R. Relaxation-Based Magic-Angle Spinning NMR Approaches for Studying Protein Dynamics eMagRes (2016) 5(3), 1423–1434. DOI: 10.1002/9780470034590.emrstm1417
|
|
Solid-state magic angle spinning (MAS) NMR relaxation measurements provide a powerful tool for characterizing protein dynamics. We provide a general introduction to contemporary relaxation-based MAS NMR approaches by reviewing the basic theoretical concepts, experimental considerations, and example procedures for quantitative analysis. We discuss methods suitable for characterization of motions occurring on time scales in the range from picoseconds to milliseconds. We also sketch out strategies to obtain time scales, amplitudes, and directions of molecular motions. |

30. Hoop , C.L., Lin, H.-K., Kar, K., Magyarfalvi, G. Lamley, J.M., Boatz, J.C., Mandal, A., Lewandowski, J.R., Wetzel, R., van der Wel P.C.A. Huntingtin exon 1 fibrils feature an interdigitated β-hairpin–based polyglutamine core PNAS (2016) 113(6) 1546–1551. DOI: 10.1073/pnas.1521933113
Accepted version of the article is freely available from WRAP: http://wrap.warwick.ac.uk/76070/
 |
Polyglutamine expansion within the exon1 of huntingtin leads to protein misfolding, aggregation, and cytotoxicity in Huntington’s Disease. This incurable neurodegenerative disease is the most prevalent member of a family of CAG repeat expansion disorders. Although mature exon1 fibrils are viable candidates for the toxic species, their molecular structure and how they form have remained poorly understood. Using advanced magic angle spinning solid state NMR, we directly probe the structure of the rigid core that is at the heart of huntingtin exon1 fibrils and other polyglutamine aggregates, via measurements of long-range intra- and inter-molecular contacts, backbone and side chain torsion angles, relaxation measurements, and calculations of chemical shifts. These reveal the presence of β-hairpin-containing β-sheets that are connected through interdigitating extended side chains. Despite dramatic differences in aggregation behavior, huntingtin exon1 fibrils and other polyglutamine-based aggregates contain identical β- strand-based cores. Prior structural models, derived from X-ray fiber diffraction and computational analyses, are shown to be inconsistent with the solid-state NMR results. Internally, the polyglutamine amyloid fibrils are co-assembled from differently structured monomers, which we describe as a type of ‘intrinsic’ polymorphism. A stochastic polyglutamine-specific aggregation mechanism is introduced to explain this phenomenon. We show that the aggregation of mutant huntingtin exon1 proceeds via an intramolecular collapse of the expanded polyglutamine domain, and discuss the implications of this observation for our understanding of its misfolding and aggregation mechanisms. |
2015| Jump to top
29. VIP: Lamley, J.M., Öster, C., Stevens, R.A., Lewandowski, J.R. Intermolecular interactions and protein dynamics by SSNMR Angewandte Chemie (2015) 54(51), 15374–15378. DOI: 10.1002/anie.201509168
Open Access Article. Published version also freely available from WRAP: http://wrap.warwick.ac.uk/id/eprint/73941
Supporting information (with corrected typo in the coeffcient for NH R1ρ): ![]()
 1521-3773/homepage/2002_vip.gif)
|
|
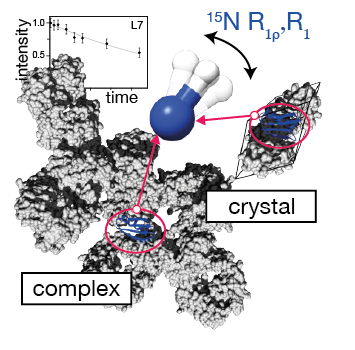 |
Understanding the dynamics of interacting proteins is a crucial step toward describing many biophysical processes. Here we investigate the backbone dynamics for protein GB1 in two different assemblies: crystalline GB1 and precipitated >300 kDa GB1-antibody complex. We perform these measurements on samples containing as little as 8 nanomoles of protein. From measurements of site-specific 15N relaxation rates including relaxation dispersion we obtain snapshots of dynamics spanning nine orders of magnitude in terms of time scale. Comparison of measurements for GB1 in either environment reveals that while many of the dynamic features of the protein are conserved between them (in particular for the fast (ps-ns) motions), much greater differences occur for slow motions with >500 ns range motions being more prevalent in the complex. The data suggest that GB1 can potentially undergo a small-amplitude overall anisotropic motion sampling the interaction interface in the complex. |
28. Lamley, J.M., Lougher, M.J., Sass, H.J., Rogowski, M., Grzesiek, S., Lewandowski, J.R. Unraveling the complexity of protein backbone dynamics with combined 13C and 15N solid-state NMR relaxation measurements PCCP 17(34), 21997-22008. DOI: 10.1039/C5CP03484A
Open Access Article. Published version also freely available from WRAP: http://wrap.warwick.ac.uk/id/eprint/73677
Correction: There is an easy to spot but egregious typographical/copy-paste error propagated throughout the formulas in the paper: the Planck's constant should be obviously in the numerator rather than denominator for the dipolar coupling related coefficients (eq. 3 & 5 in the paper and eq. 6, 8-12, 15-18). Just to confirm the correct formulas were actually used for the analysis.
Here is the version of the SI with the correct expressions: SI
|
|
|
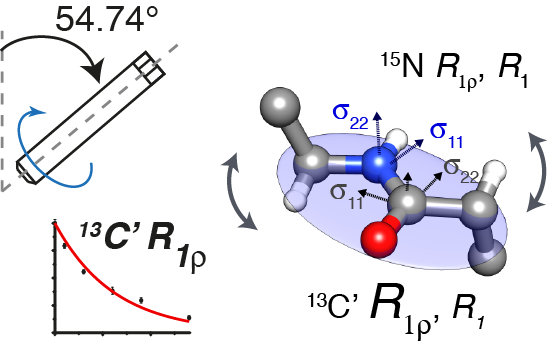 |
Typically, protein dynamics involve a complex hierarchy of motions occurring on different time scales between conformations separated by a range of different energy barriers. NMR relaxation can in principle provide a site-specific picture of both the time scales and amplitudes of these motions, but independent relaxation rates sensitive to fluctuations in different time scale ranges are required to obtain a faithful representation of the underlying dynamic complexity. This is especially pertinent for relaxation measurements in the solid state, which report on dynamics in a broader window of time scales by more than 3 orders of magnitudes compared to solution NMR relaxation. To aid in unraveling the intricacies of biomolecular dynamics we introduce 13C spin-lattice relaxation in the rotating frame (R1ρ) as a probe of backbone nanosecond-microsecond motions in proteins in the solid state. We present measurements of 13C’ R1ρ rates in fully protonated crystalline protein GB1 at 600 and 850 MHz 1H Larmor frequencies and compare them to 13C’ R1, 15N R1 and R1ρ measured under the same conditions. The addition of carbon relaxation data to the model free analysis of nitrogen relaxation data leads to greatly improved characterization of time scales of protein backbone motions, minimizing the occurrence of fitting artifacts that may be present when 15N data is used alone. We also discuss how internal motions characterized by different time scales contribute to 15N and 13C relaxation rates in the solid state and solution state, leading to fundamental differences between them, as well as phenomena such as underestimation of picosecond-range motions in the solid state and nanosecond-range motions in solution. |
27. Lewandowski, J.R., Halse, M.E.; Blackledge, M., Emsley, L. Direct observation of hierarchical protein dynamics Science 348 (6234), 578-581 (2015). http://www.sciencemag.org/content/348/6234/578 Accepted version of the article is freely available from WRAP: http://wrap.warwick.ac.uk/id/eprint/71583
|
|
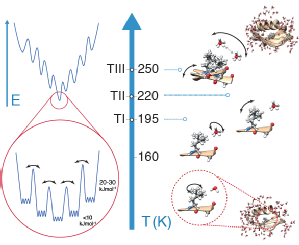 |
One of the fundamental challenges of physical biology is to understand the relationship between protein dynamics and function. At physiological temperatures, functional motions arise from the complex interplay of thermal motions of proteins and their environments. Here, we determine the hierarchy in the protein conformational energy landscape that underlies these motions, based on a series of temperature-dependent magic-angle spinning multinuclear nuclear-magnetic-resonance relaxation measurements in a hydrated nanocrystalline protein. The results support strong coupling between protein and solvent dynamics above 160 kelvin, with fast solvent motions, slow protein side-chain motions, and fast protein backbone motions being activated consecutively. Low activation energy, small-amplitude local motions dominate at low temperatures, with larger-amplitude, anisotropic, and functionally relevant motions involving entire peptide units becoming dominant at temperatures above 220 kelvin. |
2014| Jump to top
26. JACS Spotlights article: Lamley, J.M., Iuga, D., Öster, C., Sass, H.J., Rogowski, M., Oss, A., Past, J., Reinhold, A., Grzesiek, S., Samoson, A., Lewandowski, J.R. Solid-State NMR of a Protein in a Precipitated Complex with a Full-Length Antibody J. Am. Chem. Soc. 136, 16800-16806 (2014). http://pubs.acs.org/doi/abs/10.1021/ja5069992
Open Access publication. Published version of the article also freely available from WRAP: http://wrap.warwick.ac.uk/id/eprint/66503
![]()
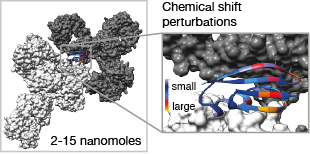 |
We demonstrate that the application of 1H-detected experiments at magic-angle spinning frequencies of >50 kHz enables the recording, in a matter of minutes to hours, of solid-state NMR spectra suitable for quantitative analysis of protein complexes present in quantities as small as a few nanomoles (tens of micrograms for the observed component). This approach enables direct structure determination and quantitative dynamics measurements in domains of protein complexes with masses of hundreds of kilodaltons. Protein–protein interaction interfaces can be mapped out by comparison of the chemical shifts of proteins within solid-state complexes with those of the same constituent proteins free in solution. We employed this methodology to characterize a >300 kDa complex of GB1 with full-length human immunoglobulin, where we found that sample preparation by simple precipitation yields spectra of exceptional quality, a feature that is likely to be shared with some other precipitating complexes. Finally, we investigated extensions of our methodology to spinning frequencies of up to 100 kHz. |
25. JACS Spotllights article: Good, D.B., Wang, S., Ward, M.E., Struppe, J.O., Brown, L.S., Lewandowski, J.R., Ladizhansky, V. Conformational dynamics of a seven transmebrane helical protein Anabaena Sensory Rhodopsin probed by solid-state NMR. J. Am. Chem. Soc. 136, 2833–2842 (2014). http://pubs.acs.org/doi/abs/10.1021/ja411633w
Accepted version of the manuscript freely available from WRAP: http://wrap.warwick.ac.uk/62548/
![]()
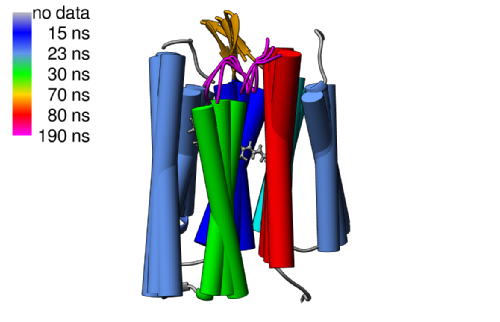 |
In this study, we report solid-state NMR site-specific measurements of the dipolar order parameters and 15N rotating frame spin–lattice (R1ρ) relaxation rates in a seven transmembrane helical protein Anabaena Sensory Rhodopsin reconstituted in lipids. The magnitudes of the observed order parameters indicate that both the well-defined transmembrane regions and the less structured intramembrane loops undergo restricted submicrosecond time scale motions. In contrast, the R1ρ rates, which were measured under fast magic angle spinning conditions, vary by an order of magnitude between the TM and exposed regions and suggest the presence of intermediate time scale motions. Using a simple model, which assumes a single exponential autocorrelation function, we estimated the time scales of dominant stochastic motions to be on the order of low tens of nanoseconds for most residues within the TM helices and tens to hundreds of nanoseconds for the extracellular B–C and F–G loops. These relatively slow time scales could be attributed to collective anisotropic motions. We used the 3D Gaussian axial fluctuations model to estimate amplitudes, directions, and time scales of overall motions for helices and the extracellular B–C and F–G loops. Within this model, the TM helices A,B,C,D,E,F undergo rigid body motions on a time scale of tens of nanoseconds, while the time scale for the seventh helix G approaches 100 ns. Similar time scales of roughly 100–200 ns are estimated for the B–C and F–G loops. The manuscript was featured in JACSSelect: Protein Dynamics in Simulation and Experiment |
2013| Jump to top
24. Lewandowski, J.R. Advances in Solid-State Relaxation Methodology for Probing Site-Specific Protein Dynamics. Acc. Chem. Res. 46, 2018-27 (2013). DOI
 |
In this review I discuss recent developments in solid-state NMR relaxation methodology for probing site-specific protein dynamics. |
2012| Jump to top
23. Mollica, L. Baias, M., Lewandowski, J.R., Wylie, B.J., Sperling, L.J., Rienstra, C.M., Emsley, L., Blackledge, M. Atomic-Resolution Structural Dynamics in Crystalline Proteins from NMR and Molecular Simulation. J. Phys. Chem. Lett. 3, 3657–3662 (2012). DOI
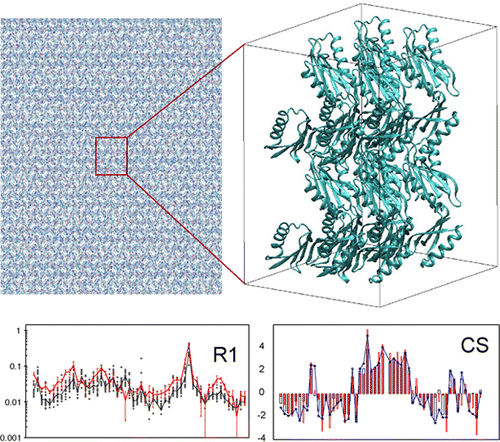 |
We compare experimentally determined dynamic parameters, spin relaxation, chemical shifts, and dipolar couplings, to values calculated from a 200 ns MD simulation of protein GB1 in its crystalline form, providing insight into the nature of structural dynamics occurring within the crystalline lattice. This simulation allows us to test the accuracy of commonly applied procedures for the interpretation of experimental solid-state relaxation data in terms of dynamic modes and time scales. |
22. Lamley, J.M. & Lewandowski, J.R. Simultaneous acquisition of homonuclear and heteronuclear long-distance contacts with time-shared third spin assisted recoupling. J. Magn. Reson. 218, 30-4 (2012). DOI
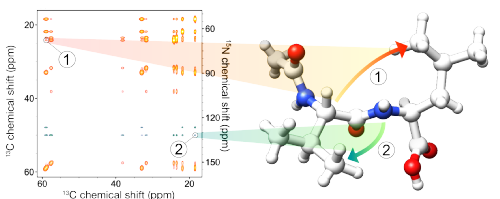 |
Time-shared Third Spin Assisted Recoupling (TSTSAR) experiment allows for simultaneous acquisition of homonuclear (13C-13C) and heteronuclear (15N-13C) long-distance contacts in biomolecular solids under magic angle spinning. TSTSAR leads to substantial time savings and increases the information content of 2D correlation spectra. |
21. Giffard, M., Hediger, S., Lewandowski, J.R., Bardet, M., Simorre, J-P., Griffin, R.G., De Paëpe, G. Compensated second-order recoupling: application to third spin assisted recoupling. Phys. Chem. Chem. Phys. 14, 7246-55 (2012). DOI
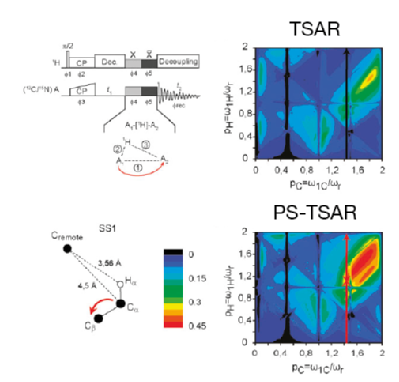 |
The modified pulse sequences of PAR and PAIN-CP involving phase inversions: Phase-Shifted Proton Assisted Recoupling (AH-PS-PAR) and Phase-Shifted Proton-Assisted Insensitive Nuclei Cross Polarization (ABH-PS-PAIN-CP) are characterized by improved polarization transfer as well as substantial broadening of the matching conditions. PS-TSAR greatly improves on the standard TSAR based methods by alleviating their sensitivity to precise RF settings and thus rendering them more robust. |
2011| Jump to top
20. Lewandowski, J.R., Sass, H.J., Grzesiek, S., Blackledge, M. & Emsley, L. Site-specific measurement of slow motions in proteins. J. Am. Chem. Soc. 133, 16762-5 (2011).
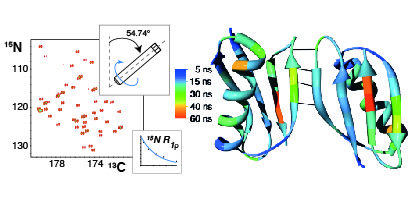 |
We demonstrate that site-specific 15N rotating-frame relaxation rates at high magic-angle-spinning frequencies provide a quantitative measure of slow protein motions in the solid state. This methodology is applicable to both perdeuterated and fully protonated protein samples. |
19. Lewandowski, J.R., van der Wel, P.C.A., Rigney, M., Grigorieff, N., Griffin, R.G. Structural Complexity of a Composite Amyloid Fibril. J. Am. Chem. Soc. 133, 14686-14698 (2011). DOI: 10.1021/ja206815h
18. De Paëpe, G., Lewandowski, J.R., Loquet, A., Eddy, M., Megy, S., Böckmann, A., Griffin, R.G. Heteronuclear proton assisted recoupling. J. Chem. Phys. 134, 095101 (2011). DOI: http://dx.doi.org/10.1063/1.3541251
17. Lewandowski, J.R., Dumez, J-N., Akbey, Ü., Lange, S., Emsley, L., Oschkinat, H. Enhanced Resolution and Coherence Lifetimes in the Solid-State NMR Spectroscopy of Perdeuterated Proteins under Ultrafast Magic-Angle Spinning. J. Phys. Chem. Lett. 2, 2205-2211 (2011). DOI: http://dx.doi.org/10.1021/jz200844n
16. Bertini I., Emsley L., Felli I.C., Laage S., Lesage A., Lewandowski J.R., Marchetti A., Pierattelli R., Pintacuda G. High-resolution and sensitivity through-bond correlations in ultra-fast magic angle spinning (MAS) solid-state NMR. Chem. Sci. 2011;2(2):345.
2010
15. Bertini, I., Bhaumik, A., De Paëpe, G.,Griffin, R.G., Lelli, M.,Lewandowski, J.R.,Luchinat, C. High-resolution solid-state NMR structure of a 17.6 kDa protein. J. Am. Chem. Soc. 132, 1032-40 (2010).
14. Barbet-Massin E., Ricagno S., Lewandowski, J.R., Giorgetti, S., Bellotti, V., Bolognesi, M., Emsley, L., Pintacuda, G. Fibrillar vs crystalline full-length beta-2-microglobulin studied by high-resolution solid-state NMR spectroscopy. J. Am. Chem. Soc. 132, 5556-7 (2010).
13. van der Wel, P.C.A., Lewandowski, J.R., Griffin, R.G. Structural characterization of GNNQQNY amyloid fibrils by magic angle spinning NMR. Biochemistry 49, 9457-69 (2010).
12. Lewandowski, J.R. et al. Measurement of site-specific 13C spin-lattice relaxation in a crystalline protein. J. Am. Chem. Soc. 132, 8252-4 (2010).
11. Lewandowski, J.R., Sein, J., Blackledge, M. & Emsley, L. Anisotropic collective motion contributes to nuclear spin relaxation in crystalline proteins. J. Am. Chem. Soc. 132, 1246-8 (2010).
2009
10. Lewandowski, J.R. De Paëpe, G., Eddy, M.T., Struppe, J., Maas, W., Griffin, R.G. Proton assisted recoupling at high spinning frequencies. J. Phys. Chem. B 113, 9062-9 (2009). DOI: http://dx.doi.org/10.1021/jp810280t
 |
We demonstrate the successful application of 13C−13C proton assisted recoupling (PAR) on [U−13C,15N] N-f-MLF-OH and [U−13C,15N] protein GB1 at high magic angle spinning (MAS) frequencies (ωr/2π = 65 kHz). Specifically, by combining PAR mixing with low power heteronuclear decoupling (ω1H/2π ∼ 16 kHz) and high spinning frequencies, we obtain high resolution 2D spectra displaying long-range 13C−13C contacts from which distance estimates can be extracted. These experiments therefore demonstrate the possibility of performing high resolution structural studies in the limit of high spinning frequency and low power 1H decoupling, a regime which optimizes the resolution of protein samples and preserves their integrity. |
9. Lewandowski, J.R., De Paëpe, G., Eddy, M.T. & Griffin, R.G. 15N-15N proton assisted recoupling in magic angle spinning NMR. J. Am. Chem. Soc. 131, 5769-76 (2009). DOI: http://dx.doi.org/10.1021/ja806578y
 |
We describe a new magic angle spinning (MAS) NMR experiment for obtaining 15N−15N correlation spectra. The approach yields direct information about the secondary and tertiary structure of proteins, including identification of α-helical stretches and interstrand connectivity in antiparallel β-sheets, which are of major interest for structural studies of membrane proteins and amyloid fibrils. The method, 15N−15N proton assisted recoupling (PAR), relies on a second-order mechanism, third spin assisted recoupling (TSAR), used previously in the context of 15N−13C and 13C−13C polarization transfer schemes. In comparison to 15N−15N proton-driven spin diffusion experiments, the PAR technique accelerates polarization transfer between 15N’s by a factor of ∼102−103 and is furthermore applicable over the entire range of currently available MAS frequencies (10−70 kHz). |
2008
8. De Paëpe, G., Lewandowski, J.R., Loquet, A., Böckmann, A. & Griffin, R.G. Proton assisted recoupling and protein structure determination. J. Chem. Phys. 129, 245101 (2008). DOI: http://dx.doi.org/10.1063/1.3036928
7. De Paëpe, G., Lewandowski, J.R. & Griffin, R.G. Spin dynamics in the modulation frame: application to homonuclear recoupling in magic angle spinning solid-state NMR. J. Chem. Phys. 128, 124503 (2008).
 |
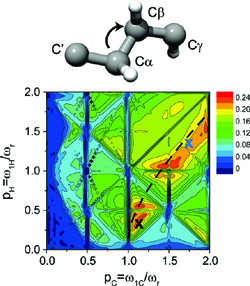
We introduce a family of solid-state NMR pulse sequences that generalizes the concept of second averaging in the modulation frame and therefore provides a new approach to perform magic angle spinning dipolar recoupling experiments. Here, we focus on two particular recoupling mechanisms—cosine modulated rotary resonance CMpRR and cosine modulated recoupling with isotropic chemical shift reintroduction COMICS. The first technique, CMpRR, is based on a cosine modulation of the rf phase and yields broadband double-quantum DQ 13C recoupling using 70 kHz ω1,C/2π rf field for the spinning frequency ωr/2π=10–30 kHz and 1H Larmor frequency ω0,H /2π up to 900 MHz. Importantly, for p ≤ 5, CMpRR recouples efficiently in the absence of 1H decoupling. Extension to lower p values 3.5 ≤p ≤5 and higher spinning frequencies is possible using low power 1H irradiation 0.25ωr/2π. This phenomenon is explained through higher order cross terms including a homonuclear third spin assisted recoupling mechanism among protons. CMpRR mitigates the heating effects of simultaneous high power 13C recoupling and 1H decoupling. The second technique, COMICS, involves low power 13C irradiation that induces simultaneous recoupling of the 13C DQ dipolar and isotropic chemical shift terms. In contrast to CMpRR, where the DQ bandwidth 30 kHz at ω0,H/2π=750 MHz covers the entire 13C spectral width, COMICS recoupling, through the reintroduction of the isotropic chemical shift, is selective with respect to the carrier frequency, having a typical bandwidth of 100 Hz. This approach is intended as a general frequency selective method circumventing dipolar truncation supplementary to R2 experiments. These new γ-encoded sequences with attenuated rf requirements extend the applicability of homonuclear recoupling techniques to new regimes—high spinning and Larmor frequencies—and therefore should be of major interest for high resolution biomolecular studies. |
2007
6. Lewandowski, J.R., De Paëpe, G. & Griffin, R.G. Proton assisted insensitive nuclei cross polarization. J. Am. Chem. Soc. 129, 728-9 (2007).
5. van der Wel, P.C.A., Lewandowski, J.R. & Griffin, R.G. Solid-state NMR study of amyloid nanocrystals and fibrils formed by the peptide GNNQQNY from yeast prion protein Sup35p. J. Am. Chem. Soc. 129, 5117-30 (2007).
2006
4. Ramachandran, R., Lewandowski, J.R., van der Wel, P.C.A. & Griffin, R.G. Multipole-multimode Floquet theory of rotational resonance width experiments: 13C-13C distance measurements in uniformly labeled solids. J. Chem. Phys. 124, 214107 (2006).
3. van der Wel, P.C.A., Hu, K.-N., Lewandowski, J.R. & Griffin, R.G. Dynamic nuclear polarization of amyloidogenic peptide nanocrystals: GNNQQNY, a core segment of the yeast prion protein Sup35p. J. Am. Chem. Soc. 128, 10840-6 (2006).
2. De Paëpe, G., Lewandowski, J.R., Bayro, M.J. & Griffin, R.G. Broadband homonuclear correlation spectroscopy at high magnetic fields and MAS frequencies. J. Am. Chem. Soc. 128, 1776-7 (2006).
2001
1. Pecul, M., Lewandowski, J.R. & Sadlej, J. Benchmark calculations of the shielding constants in the water dimer. Chem. Phys. Lett. 333, 139-145 (2001).