
ThermoPhyl Users guide
This manual provides a quick working guide to ThermoPhyl, a pattern-matching script that rapidly compares and summarizes the number of matches between a list of candidate assays and designated target and non-target sequences. ThermoPhyl is designed for applications in which a user wishes to specifically and sensitively target a taxonomic group of interest in a complex sample using quantitative-PCR or traditional PCR. The name ThermoPhyl derives from its central goal which is to test thermodynamically optimal PCR assays for phylogenetic sensitivity and specificity to arrive at an assay which is both thermodynamically and phylogenetically optimal. In the past, users have typically designed PCR primers and/or probes manually, by visual comparison against multiple alignment files. Even when a phylogenetically suitable assay (i.e. maximally sensitive and specific) can be determined, empirical tests are laborious and often produce poor PCR results.
ThermoPhyl turns this process around by starting with candidate assays which should all produce an efficient PCR. ThermoPhyl is designed to use as input a very large number of candidate assays and assess each one for phylogenetic sensitivity and specificity. The outputs are summary tables of the number of matches to sequences designated as target and non-target groups by the user.
Installing and Running ThermoPhyl
Before ThermoPhyl can be run, PERL must be installed. A download is available.
To install ThermoPhyl, the program file simply needs to be copied into the PERL directory or, if PERL is added to the computer’s path, any directory of choice.
To run ThermoPhyl, just execute the script whether in a Unix or Windows environment. In Windows, double-click on the file, or call up a command window (Start->Run->’cmd’), navigate to the directory where you have copied ThermoPhyl (for example, ‘cd C:\PERL’) and type the filename to start the program.
ThermoPhyl requires three input files:
-
A fasta file which contains all of the target and non-target sequences you wish to test (has to be called “outside_world.fas”). The more sequences this file contains, the higher the confidence in distinguishing between target and non-target groups. This file can contain the users own sequences and/or sequences retrieved from public databases. Typically, this file might contain 100 – 50,000 sequences. Be aware that many databases such as GreenGenes and Silva for 16S rRNA genes contain many very similar sequences; users will generally want to reduce these databases to some sort of core set of representative sequences.
-
A text file containing only the names of target sequences (has to be called “target_list.txt”). The names must correspond to those in the fasta file above and should be some sort of unique identifier, like a GenBank Accession number or GreenGenesID. A column heading can be present or not.
-
A list of candidate assays for traditional PCR or qPCR (has to be called “candidate_assays.txt”). The recommended approach is to first compile all target sequences in a single fasta file. For traditional PCR, this file can be read directly into BatchPrimer3 (https://probes.pw.usda.gov/cgi-bin/batchprimer3/batchprimer3.cgi) or for qPCR, each sequence can be input into ABI’s PrimerExpress to generate a list of candidate assays to test. We typically generate 50 candidate assays per target sequence. All possible candidate assays should be compiled into a single tab-delimited text file (.txt) with columns in the order of Forward Primer, Probe (if present), Reverse Primer, with or without column headings. The file can contain >10,000 candidate assays and should look like this:
FORWARD PROBE REVERSE
TGATTGACCACACCCGTATTACC GCCGTTCACCTCAGCCTTAG ATCTCTGCTTGTCCGCTC
CGCTGTTCATGCTTCCGATA GATCGATCATCGGCGGTTT CCTCGGTGTGCATCG
Or like this for Primer3 output:
TGATTGACCACACCCGTATTACC
ATCTCTGCTTGTCCGCTC
CGCTGTTCATGCTTCCGATA
CCTCGGTGTGCATCG
In the last example of output, the 1st sequence in the list is a forward primer, the 2nd the matching reverse primer, and so on. Each line should have only a line return (paragraph mark) at the end and in the case of multiple columns, each column should be tab-delimited.
Specific Recommendations and Potential Pitfalls:
-
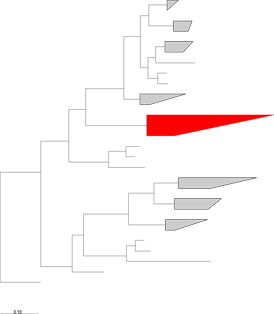
Target groups should form some sort of natural phylogenetic group. Sequences should be properly placed in some sort of a phylogenetic tree to evaluate this and to designate target and non-target sequences in a way which reflects the evolutionary history of the genetic locus or loci in question. In the cartoon to the left, sensitive and specific assays could probably be designed to successfully distinguish between sequences in the red clade and other, non-target, taxa in the rest of the tree. If your target sequences do not form a coherent phylogenetic group, it will obviously be difficult to design an accurate assay, although it is possible that different sequence data (i.e. different portions of the same alignment or another locus) for the same taxa could still be used in such a case.
-
The more sequence data available for both target and non-target groups, the better. The strength of ThermoPhyl, in fact its central goal, is to summarize a very large number of comparisons to arrive at a single ‘best’ assay. Particularly with environmental samples, we almost always have an incomplete knowledge base, and so may be blind to the best assay.
-
Files must be in the formats described above. Common problems are listed in the FAQ.