
(Inter)facing the Bitter Truth: How to Design Better Interfaces in Next-Gen Batteries using Atomistic Simulations Assisted by Machine-Learning
Supervisors: Bora Karasulu and Albert Bartok-Partay
Supervisors: Bora Karasulu and Albert Bartok-Partay
Summary:
Lithium-Sulphur batteries (LSBs) are a promising alternative to Li-ion batteries (LIBs) as a next-gen energy storage technology, providing higher theoretical capacity at lower costs. Replacing the conventional liquid electrolytes with solid electrolytes (SE) helps mitigate the major LSB issues like the Li-polysulfide shuttle effect, and safety risks. Current SEs, however, degrade when coupled with a S-cathode, impeding the Li-ion conduction across their interfaces, limiting the battery performance. To design superior SE/S-cathode interfaces, this project focuses on atomistic simulations of the interfacial sulphide conversion chemistry in LSBs utilising state-of-the-art Density Functional Theory and machine learning methods, providing insights that are otherwise elusive to experimental characterisation techniques.
Background:
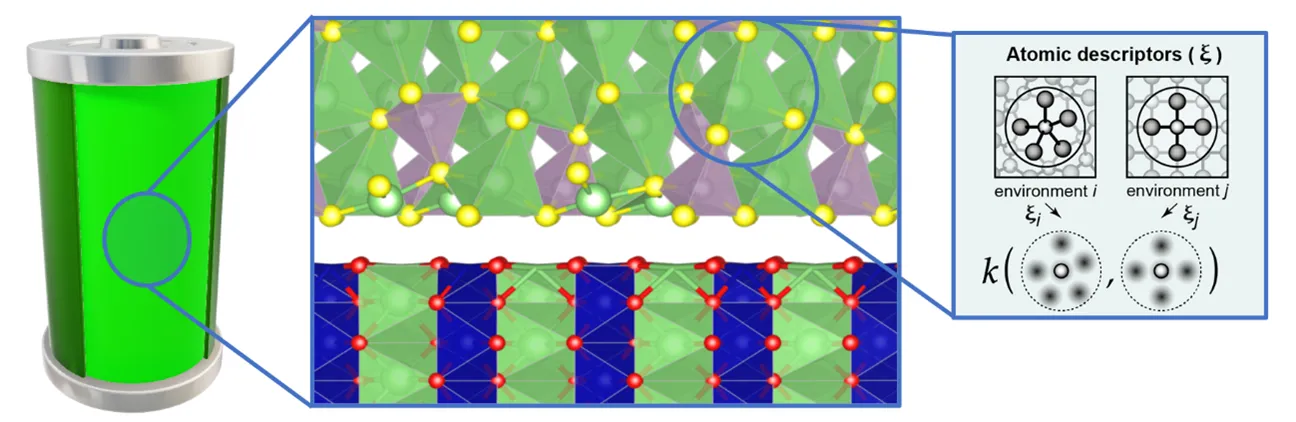
Li-S batteries (LSBs) are a promising alternative to Li-ion batteries (LIBs) as a next-gen energy storage technology, owing to their very high theoretical capacity and low cost [1]. In an all-solid-state battery (ASSB), a solid electrolyte (SE) replaces the traditional liquid electrolyte, mitigating the related issues like the shuttle effect of Li-polysulfides, and safety risks. However, various SEs are known to degrade when interfaced with a S-cathode, forming polysulphides that impede the Li-ion conduction across interfaces and the cathode overpotential, limiting the battery performance. To design superior SE-cathode interfaces, the interfacial sulphide conversion chemistry must be known, but experimental characterisation using tools like SEM and TEM is challenging due to the volatility of sulphur [2], rendering atomistic simulations a viable recourse.
Ab initio (DFT) methods are routinely used to discover and characterise bulk ASSB materials, but their applications in modelling interfaces are rather limited, mainly due to the much higher computational costs [3]. Larger models are needed to simulate interfaces that adequately retain the bulk properties and minimise the artificial lattice strain between the two surfaces. Also, longer simulations (>100ps) are vital in sampling the Li dendrite growth and polysulphide formation processes. Scaling-up requires ML interatomic potentials (MLIP), that provide near-DFT accuracies at a fraction of DFT costs.
This project therefore focuses on atomistic simulations of the SE-cathode interfaces within LSBs under charging/discharging conditions, representing changes in the chemical states and bonding of the particles. The presence of particles whose oxidation state changes during the simulations requires explicit treatment of electrostatics within the MLIP framework, calling for the extension of current ML models. Therefore, in the project the PhD student will also develop novel MLIP frameworks.
Links to HetSys Training
The HetSys training programme will introduce the student to appropriate background via core modules, covering quantum mechanics and atomistic simulations as well as machine learning (ML) and uncertainty quantification. The student will have the opportunity to study additional modules relevant to the project, such as electronic structure theory and quantum chemistry. The bespoke software carpentry training will enable the student to work efficiently on code development and implementation.Proposed work spans materials chemistry, electrochemistry, solid-state chemistry and physics, and highly synergetic with experimental work. The PhD candidate will present results at frequent interdisciplinary catch-up meetings between theory and experimental teams, giving them experience in communicateing challenging concepts effectively.
Machine learning potentials will be based on Bayesian fitting methods, providing uncertainty estimates, which will be propagated through the simulation pipeline to provide uncertainty estimates of the predicted thermodynamic properties. Statistical uncertainties are rigorously quantified within the framework.
Robust and reproducible workflows will be used to carry out complex calculations involving multiple software packages. These include DFT, structure search methods, ML model fitting as well as molecular dynamics codes. There will be methodological and software development to implement long-range electrostatic interactions.
References
[1] Energy Fuels 2020, 34, 10, 11942–61;
[2] Power Sources 2016, 319, 247– 54;
[3] Prog. Energy 2022, 4 012002