
Protein origami: New computational methods to predict protein folding ensembles
Student: Ziad Fakhoury
Supervisors: Scott Habershon, Gabriele Sosso
How does a protein fold into its native state? This is one of the most important and challenging problems in the chemical sciences, and a key question in understanding diseases driven by protein misfolding and aggregation (such as Parkinson’s disease). In this project, we will develop and employ a new computational scheme to access long time-scale protein folding events by mapping onto a discretized connectivity-based description of protein structure. This new approach will then enable us to investigate sequence-specific folding effects and translational folding, as well as providing a new scheme for protein-structure prediction.
Predicting protein-folding pathways is challenging because of the timescales involved; folding usually takes micro- to milliseconds, a regime which is far beyond the reach of normal molecular simulations. Furthermore, protein folding is strongly coupled to the solvent environment (e.g. water), demanding new approaches which can access long timescales simulations in heterogeneous solvent/protein environments
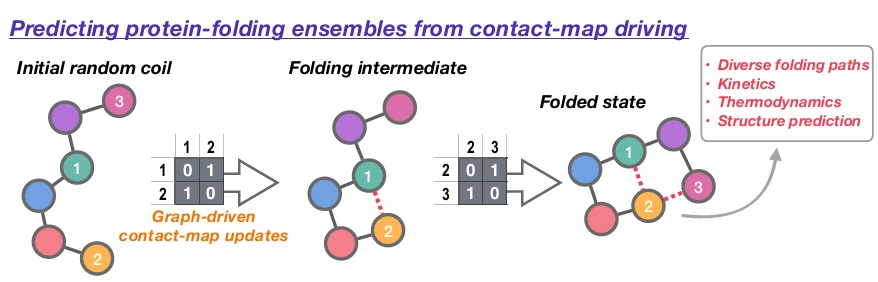
To address this frontier challenge, we will develop a new computational scheme to predict how proteins fold into their native states. Building on our previous work in the field of reaction discovery, protein folding pathways will be encoded as a series of “hops” between discrete intermediate structures defined using residue-level adjacency matrices; based on a matrix-space search introduced in our recent work, ensembles of folding mechanisms can be readily generated within this discretised folding picture, while post-analysis of thermodynamic and kinetic descriptors for different pathways enables identification of the most likely folding mechanism(s). As well as delivering new open software for generating folding-mechanism ensemble, our new approach will uniquely enable us to investigate (i) sequence-specific folding effects, (ii) how the protein-folding landscape evolves during synthesis, and (iii) how knowledge of folding mechanism can inform prediction of stable native structures.