
Complete thermodynamic description of alloys to extreme pressure and temperature
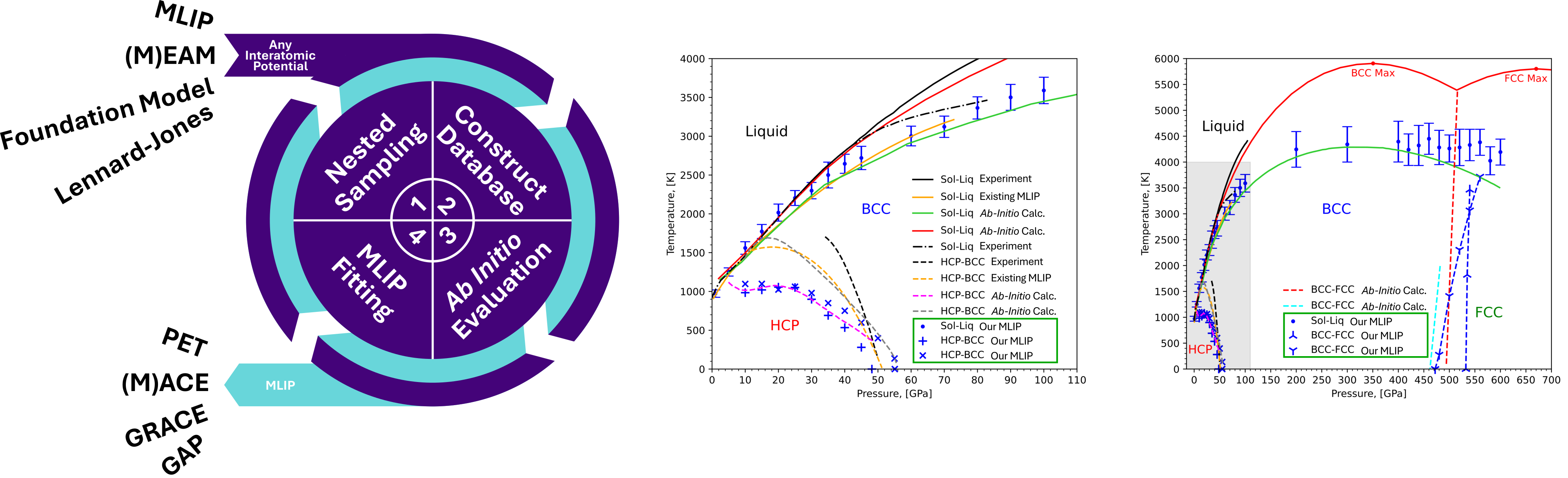
Left: Illustration of the automatic database generation procedure, devised from this PhD, which we used to train a MLIP that is able to reproduce the phase diagram of pure magnesium up to 600 GPa (centre and right) compared to experimental results (available up to ~100GPa) and pure ab-initio results (available sparingly up to 600 GPa). Illustration was reproduced from our publication.1
Summary
Machine learning interatomic potentials (MLIPs) have revolutionised the field of atomistic simulation, allowing ab-initio level accuracy force and energy predictions at a fraction of the computational cost of exact evaluations from quantum mechanics packages. The accuracy of a MLIP depends on two aspects: the model architecture and the training database. While there is plenty of understanding on how to select model architectures, there is no standardised process for generating a training database. Additionally, many current database generation schemes rely on preconceived information about the system of study, leading to ambiguity about how data points should be weighted when training a model.
As fusion reactors become closer to reality and space becomes ever more accessible, the need for materials that operate under extreme pressures and temperatures is greater than ever. Thermodynamic calculations under extreme conditions require extensive sampling, which makes ab-initio based simulations computationally unfeasible. Here MLIPs are critical for gaining an accurate understanding of a materials properties under these conditions, but without extensive experimental guidance, building MLIP databases is highly challenging. This problem is compounded by the complex potential energy surfaces created by multi-component alloy systems.
The goal of this project is to leverage atomistic sampling techniques to develop an automated scheme to construct optimal databases to train MLIPs, to allow ab-initio level thermodynamic predictions of unseen materials at experimentally inaccessible conditions.
Student: Vincent FletcherLink opens in a new window
Supervisors: Albert P. BartókLink opens in a new window and Livia B. PártayLink opens in a new window
References
Key Words
Machine Learning, Database Generation, Generative Learning, Gaussian Approximation Potentials (GAP), Atomic Cluster Expansion (ACE), Density Functional Theory (DFT), Ab-Initio, Nested Sampling, Phase Diagrams, Crystal Structures, High Performance Computing (HPC), Thermodynamics, Statistical Mechanics, Chemistry, Physics, Programming