
Untangling disorder in amorphous pharmaceuticals using artificial intelligence
Student: Jeremy Thorn
Supervisors: Albert Bartok-Partay, Steven Brown
The best pharmacologically active compound may have been found, but its function, properties and processability are heavily dependent on the solid form. Solid-state Nuclear Magnetic Resonance and diffraction experiments, coupled with quantum mechanical calculations are powerful tools to elucidate the atomic structure, which is needed to understand and control macroscopic properties. The challenge in amorphous systems is that experiments only provide globally averaged measurements, while large length scales are needed to capture the disorder, which render quantum mechanical calculations unfeasible. In this project, machine learning models will be used to bypass the need for these costly computations, allowing interpretation of experimental data and structure determination with unprecedented accuracy.
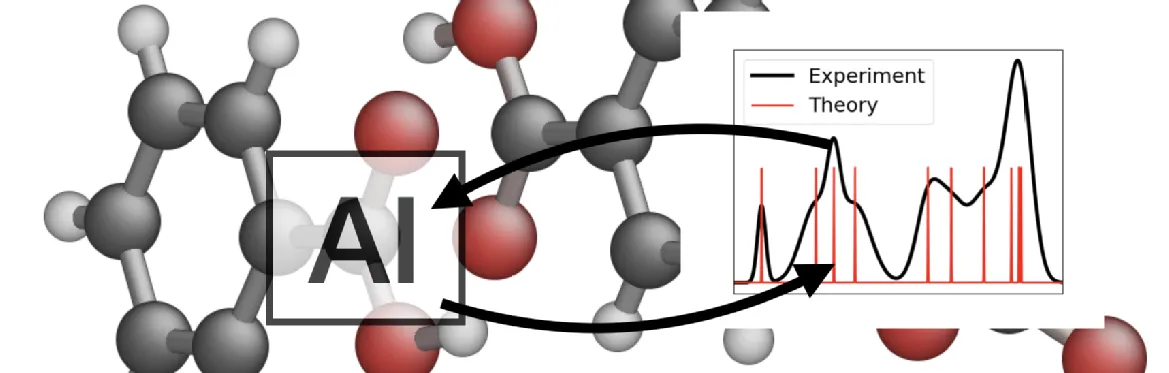
Due to their strong dependence on the local atomic environment, chemical shifts in solid-state NMR are amongst the most powerful tools for the structure elucidation of powdered solids or amorphous materials. Assigning experimentally determined chemical shifts to local structures is often challenging in the solid state, and ab initio calculations are crucial to establish the link. The Density Functional Theory based Gauge Including Projector Augmented Waves method provides great accuracy, but comes at a high computational cost, making it infeasible for extended systems. Machine learning has emerged as a way to overcome the need for quantum chemical calculations of solid-state NMR chemical shifts. It has been shown by Paruzzo et al [1] that a machine learning approach, based on local environments, is able to accurately predict the chemical shifts of molecular solids and their polymorphs.
In collaboration with AstraZeneca, we propose to exploit the expertise in solid-state NMR and machine learning at Warwick to develop a new framework for predicting solid state NMR chemical shifts and material properties for both crystalline and amorphous systems, highly relevant in the pharmaceutical industry. Local order or disorder present within a molecular material, whether crystalline or amorphous, can have an enormous impact on material properties and processability. By correlating experimentally observed changes in solid-state NMR chemical shifts with machine learning derived predictions at the atomic length scale will allow the link between atomistic changes in local order and product performance to be established. Applied in the development of pharmaceuticals, this will enable exploration of differences between batches and the impact of storage conditions on local atomic order, therefore aid devising control strategies to mitigate risk.
[1] Paruzzo, FM et al, Nat Commun 9, 4501 (2018)