
Machine-learning quantum surrogate models to simulate energy transport across interfaces
Supervisors: Reinhard Maurer and James Kermode
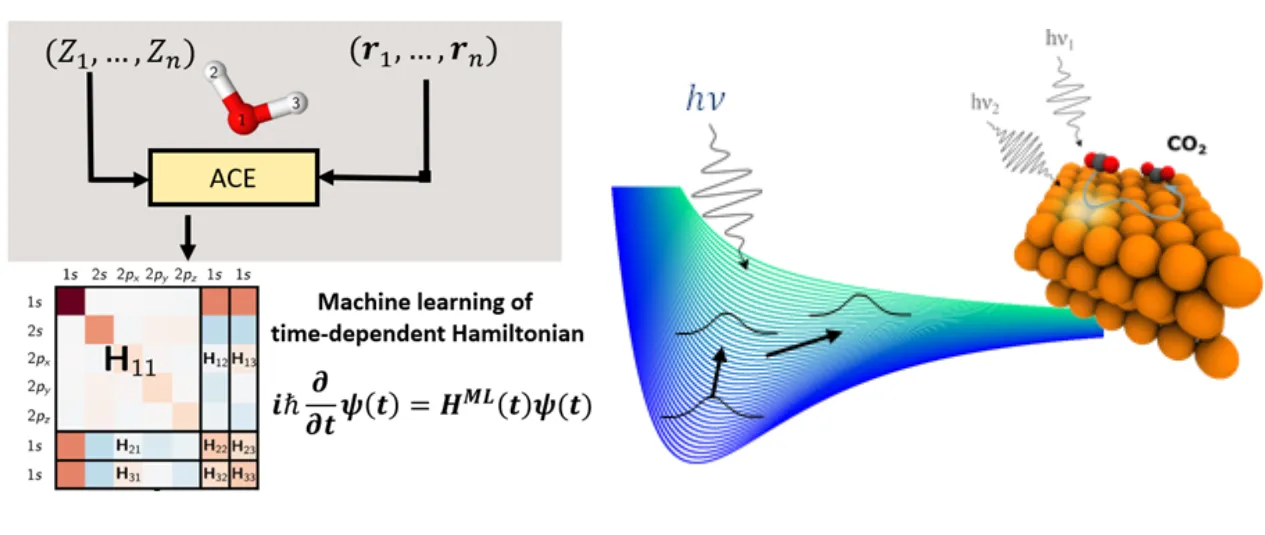
Figure:(left) Ultrafast dynamics at surfaces in lithography and catalysis involve light, electronic excitation and fast atomic motion driven by coupled electron-nuclear dynamics. (right) To make the time-dependent simulation of such ultrafast processes at the scale of tens of thousands of atoms feasible, Atomic Cluster Expansion (ACE) representations will be used to construct a quantum Hamiltonian surrogate model.
Supervisors: Reinhard Maurer and James Kermode
Summary:
Modern technologies such as photocatalysis or laser nanolithography involve energy transfer across interfaces. Many critical societal challenges require that we transfer light or electronic energy more efficiently into chemical energy, e.g., to utilize CO2 as renewable fuel. To achieve this, we need to understand the mechanisms behind the intricate dynamics that unfold at interfaces. Quantum mechanical simulations provide electronic-structure insights but are computationally intractable for relevant systems. The aim of this project is to create and apply machine learning models that emulate the quantum mechanical interaction of light, electrons, and atoms for many thousands of atoms at realistic interfaces.
Background:
Experimental evidence on ultrafast dynamics and transport at surfaces is notoriously hard to interpret on its own without knowledge of the nanoscale structure and electronic properties that unfold during the dynamics. First principles electronic structure methods are unable to address complex non-equilibrium dynamics at the scale of thousands to tens of thousands of atoms.
Machine learning (ML) methods are revolutionising the physical sciences, for example interatomic potential representations are becoming a common approach to accelerate molecular dynamics simulations. It was recently shown that ML methods can even reconstruct quantum mechanical Hamiltonians of molecules [1] and provide models of multiple electronic states [2]. ML-based surrogate models need to be able to replicate electronic structure results with a precision of a few meV to be reliable.
Research Question for the PhD project:
· Is the ACEhamiltonians representation [3] able to capture the electronic structure and energy landscape of multicomponent interfaces and nanostructures?
· How much training data and of which kind is required to generate a faithful and transferable representation of electronic structure of a material interface?
· Can we use surrogate Hamiltonians to predict measurable transport coefficients and reaction rates driven by light and temperature gradients?
In this project, you will develop a new machine learning representation of quantum mechanical Hamiltonians based on the recently proposed ACEhamiltonians approach [3]. You will expand this approach to multicomponent systems with the aim to perform large-scale non-equilibrium time-dependent simulations of ultrafast energy and charge transport at interfaces. You will generate training data based on Density Functional Theory and learn how to build such models based on the Atomic Cluster Expansion (ACE) formalism [4]. Once the models are constructed and validated, you will use a recently developed molecular dynamics simulation code [5] to apply them to simulate different dynamical processes such as light-driven defect propagation and light-driven ultrafast dynamics of molecules at surfaces.
Skills that the student will acquire:
- Machine learning and high-dimensional parametric fitting
- Electronic structure theory
- Molecular dynamics simulations and open quantum system dynamics
- Software development in Julia and Python programming languages
Links to HetSys Training:
The project spans chemistry, condensed matter physics, and mathematics. The student will need to acquire skills from all disciplines to deliver on the aims of this project.The simulations will include multiple error sources that stem from the intrinsic limitations of electronic structure theory, the errors associated with the ML representation and the error associated with the approximations inherent to the molecular dynamics simulations. The training in uncertainty quantification from PX914 will help the student to deliver robust simulation results and to carefully analyse the sensitivity of the simulations to the various error sources. The student will work on extending and supporting two existing software packages written in the Julia programming language, namely ACEhamiltonians.jl and NQCDynamics.jl for dynamics simulations. The RSE training in PX913 will provide the student with skills to contribute easily maintainable and efficient code
References:
[1] Schütt et al. „Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions“, Nature Commun. 10, 5024 (2019), https://www.nature.com/articles/s41467-019-12875-2
[2] Westermayr, Maurer, „Physically inspired deep learning of molecular excitations and photoemission spectra”, Chem. Sci. 12, 10755-10764 (2021), https://pubs.rsc.org/en/content/articlelanding/2021/sc/d1sc01542g
[3] Zhang et al. „ Equivariant analytical mapping of first principles Hamiltonians to accurate and transferable materials models”, npj Computational Materials 8, 158 (2022) https://www.nature.com/articles/s41524-022-00843-2. Implemented in ACEhamiltonians.jl package.
[4] Dusson et al. “Atomic Cluster Expansion: Completeness, Efficiency, and Stability”, J. Comput. Phys. 454, 110946 (2022), https://doi.org/10.1016/j.jcp.2022.110946
[5] Gardner et al. “NQCDynamics.jl: A Julia package for nonadiabatic quantum classical molecular dynamics in the condensed phase”, J. Chem. Phys. 156, 174801 (2022), https://pubs.acs.org/doi/abs/10.1021/acs.jctc.9b01217