Zenawi Welderufael
Personal and Project Details:
I am a PhD student at the School of Chemistry University of Southampton, funded by iMR-CDT. I am working on a project titled “Highly efficient quantum spin dynamics simulation algorithms” and dealing with biomolecular structure determination using NMR and EPR spectroscopy under the supervision of Dr. Ilya Kuprov.
Qualifications:
MSc in Advanced Spectroscopy in Chemistry, Erasmus Mundus Programme, and graduated in the 15th of June 2012.
First year at the University of Lille 1, Lille, France.
Second year at the Jagiellonian University in Krakow, Krakow, Poland.
Research:
My research focuses on large-scale simulation of NMR, EPR, and DNP experiments on macromolecular systems with the purpose of aiding NMR and EPR structure determination. The novel theoretical procedure (recently suggested by the IK group) determines atomic coordinates of molecules by direct fitting of Cartesian coordinates to the experimental spectrum. The procedure is shown schematically in Figure 1 – the initial guess atomic coordinates are used to compute magnetic properties, those are then used to compute the spectrum and the deviation from the experiment is iteratively minimized with respect to the atomic coordinates. Proof of principle calculations with this algorithm have been published, the following questions are presently open and will be the topics of my research:
1. Accuracy of the resulting structures as compared to X-Ray and traditional NMR structure determination techniques.
2. The effect of the theory level (DFT, HF, MP2, CCSD, etc.) and the basis set at the quantum chemistry stage of the algorithm on the accuracy of the resulting structure.
3. Range of applicability of the direct structure fitting algorithm – the current protein structural work is fundamentally limited to proteins, our new algorithm does not have that limitation.
4. The choice of the regularization methods – at the moment the parts of the molecule that do not influence the spectrum (such parts are often present in EPR) are optimized into a quantum chemistry energy minimum. The appropriateness of this procedure and possible alternatives will be investigated.
5. At the moment the algorithm uses the BFGS quasi-Newton minimizer with numerical gradients. Alternatives will be investigated, both in terms of speed (analytical gradients) and convergence rate (other variations of quasi-Newton methods and possibly other minimization techniques).
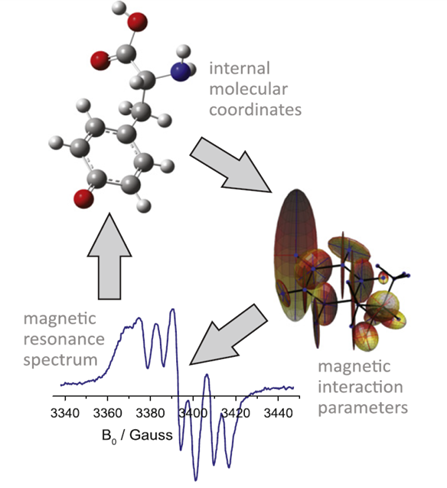
Figure 1. A schematic representation of the direct structure fitting procedure, the figure depicts that molecular coordinates provided to a quantum chemistry package and the resulting magnetic parameter are fed to a spin dynamics simulation package (spinach, developed by IK group). A squared norm of the deviation from experimental spectrum is computed, and the deviation is iteratively minimized with respect to molecular coordinates. A constraint is imposed on the molecular energy to stay within the thermally accessible energy range.
At the moment I am busy learning advanced programming techniques, spin dynamics. We are awaiting the delivery of the EPR spectrometer to the University of Southampton – some of the spectra to be fitted will be recorded by ourselves, others will be taken from databases.
A part from my recent research work on Karplus curve parameterization for the J-coupling predictor function in Protein NMR spectroscopy simulations using SPINACH. SPINACH is an open source library of algorithms for the simulation of spin dynamics in Magnetic Resonance. Magnetic parameters are computed using DFT with an exchange correlation B3LYP and basis set m06/cc-pvtz.
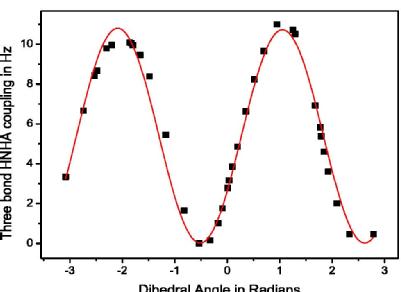
Figure 2. Fitted curve of 3J HNHα coupling constant as a function of the backbone phi dihedral angle. The fitting is achieved using Karplus equation (3J = A cos2 (φ) + B cos (φ) + C). The coefficients derived from the fitting have values of 10.73, -0.037, 0.024, and –1.052 for A, B, C, and phase respectively.
Publication:
http://dx.doi.org/10.1016/j.jmr.2014.04.002
References:
G.T.P. Charnock, M. Krzystyniak, Ilya Kuprov, Molecular structure refinement by direct fitting of atomic coordinates to experimental ESR spectra, JMR 216 (2012) 62-68.
H.J. Hogben, M. Krzystyniak, G.T.P. Charnock, P.J. Hore, I. Kuprov, Spinach - a software library for simulation of spin dynamics in large spin systems, Journal of Magnetic Resonance, 208 (2011) 179-194.

Contact
Zenawi Welderufael
Building 30/3039
University of Southampton
University Road
SOUTHAMPTON
SO17 1BJ
Tel: 02380 522391
Email: ztw1e12@soton.ac.uk
Spinach-a polynomially scaling spin dynamics simulation library