
The Correlation Threshold as a Strategy for Gene Filtering, with Application to Irritable Bowel Syndrome and Breast Cancer Microarray Data
Samuel Edward Jackson*, Jochen Einbeck*, Adetayo Kasim‡ and Willem Talloen§
*Department of Mathematical Sciences, Durham University,
‡Wolfson Research Institute, Durham University,
§Johnson & Johnson Pharmaceutical Research & Development, Division of Janssen Pharmaceutica, Beerse, Belgium
Abstract
It is well established in the literature that certain disease-associated gene signatures can be identified as a source for predicting the classification of samples or cell lines into diagnostic groups – for example, healthy and diseased. Using standard techniques for the selection of significant genes may lead to many highly correlated genes to be chosen, which may be an issue if we are limited in the number of genes we can select. This article therefore aims to investigate methods for selecting genes with the application of a correlation threshold. The methods are applied to two high-dimensional microarray datasets, one to aid the prediction of the presence or absence of Irritable Bowel Syndrome, and one to predict whether the oestrogen-receptor class of a given breast cancer cell line is positive or negative. Our results suggest that the effectiveness of the correlation threshold as a gene selection parameter depends on the particular microarray dataset and classification problem. While the correlation threshold may be beneficial in some specific scenarios where the number of required genes is restrictively small, it may also have no or even detrimental effect on the classification accuracy.
Keywords: Irritable Bowel Syndrome, microarray, gene expression, disease classification, Diagonal Linear Discriminant Analysis, correlation threshold.
Introduction
Microarray experiments have emerged to play an increasingly important role in the world of medical science. They have been used as an aid for the classification of tumours (Golub et al., 1999), the prediction of possible response to therapy and treatments (Tusher et al., 2001) and the prediction of the presence or absence of many particular diseases, including breast cancer (van't Veer et al., 2002). Microarrays measure the mRNA expression level, known as the gene expression level, for thousands of genes simultaneously (Quackenbush, 2006). The pre-regularised gene expression level of a particular gene in a particular cell indicates how much that particular gene is used in order to carry out the functions of the cell. The higher the gene expression, the more the corresponding gene proteins are used within the cell.
A typical microarray dataset consists of n subjects, which constitute the sample. Each subject has a pre-regularised measure of gene expression for each of the p genes, each gene constituting a parameter. The sample sizes, n, of gene expression microarray datasets are small compared to the number of parameters, p, that is, we have n<<p, and so standard statistical techniques should be used carefully. Measuring the gene expression value for many genes is costly. Therefore, for both financial reasons and computational efficiency, it would be invaluable to know a small handful of particularly significant genes, known as molecular biomarkers, for which the gene expression values could be used as predictors for the classification of future samples into their diagnostic groups. Then, only the expression values of these few genes need to be found in future subjects in order to make a prediction about the presence or absence of a particular disease. This will be considerably cheaper and more efficient than finding the whole genome expression values of a subject or biological sample.
An example of comparing the expression values of a more informative gene with one which is not is shown in Figure 1, which depicts the gene expression values of two particular genes from a full microarray analysis dataset comparing 34 Irritable Bowel Syndrome (IBS) patients and 24 healthy subjects. This dataset is one of those used throughout the article and is introduced in full later on. We can see that, on the whole, the more informative gene has lower expression values for the diseased group than the healthy group. In comparison, the uninformative gene has more overlapping expression value ranges for the two groups, so knowing the expression value of this gene for a further subject would give you little indication of the classification group.
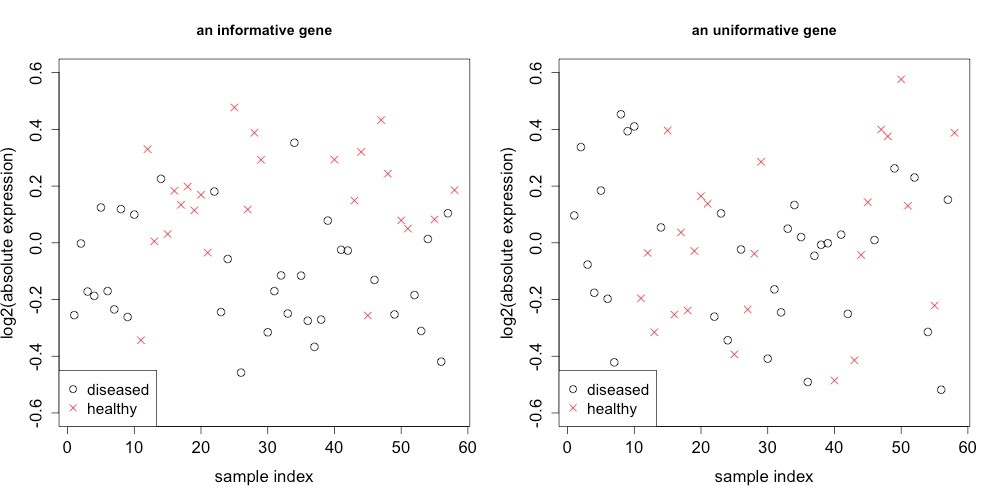
Figure 1: Comparing the plots of the expression values for a potentially informative gene and an uninformative gene, both taken from a microarray analysis dataset for IBS.
Many methods of gene selection typically involve ranking the genes with regard to a certain test statistic (Chen, 2005). A higher test statistic value for a particular gene typically corresponds to a gene being seen as more important for differentiating between two or more distinct groups – for example, healthy and diseased. If such a measure can be found, then ordering the genes with regard to decreasing test statistic value gives an order of importance. The top k genes of this ranking can then be chosen for use in classification, which is the prediction of the diagnostic category of a tissue sample from its gene expression values given the availability of similar data from tissues in identified categories (Yeung and Bumgarner, 2003). However, as mentioned by Yeung and Bumgarner (2003), and also Jaeger et al. (2003), there are problems with simply selecting the top k genes according to the ranking of a certain test statistic. Specifically, many of the genes selected will be highly correlated, and the information that can be obtained from the expression values of highly correlated genes may be very similar.
One explanation for high correlations between certain genes is that the genes all belong to one particular biological pathway (Oti and Brunner, 2007). A biological pathway is a series of actions among molecules in a cell that leads to a certain product or a change in the cell (National Human Genome Research Institute, 2016). Gene regulation pathways control the expression levels of certain sets of genes. Therefore, the gene expression values of many of the genes affected by a single pathway may change in a similar fashion. As a consequence, in order to classify the data into diagnostic groups, we may only need the expression values of one or two genes from a particular pathway to gain a similar amount of information that would be gained from having expression values for all the genes.
Additionally, if there is a limit on the number of genes which we can select, maybe due to financial cost or computational necessity, then we may not be selecting any gene from some highly influential and informative group of gene pathways. Although selecting some genes from further down the ranking list would result in genes of a lower test statistic being chosen, they may be more beneficial in combination with some of the genes that had already been chosen from near the top of the list. We therefore consider applying a correlation threshold to the gene selection process. The motivation for doing so is to remove some highly correlated, and thus possibly redundant, genes from being included when implementing the classifier.
Microarray data and pre-processing
We will be investigating the importance of the correlation threshold, and its effect on classification accuracy, using two microarray datasets. The concept of the correlation threshold itself will be explained in later sections.
The first dataset is provided by Janssen Pharmaceutica and consists of gene expression values for each of 21,212 genes for each of 34 diseased IBS patients and 24 healthy controls. IBS is a prevalent disorder affecting between 10% and 20% of people in the Western world (Aerssens et al., 2008). It is characterised by recurrent abdominal pain and an increased frequency to need to empty the bowels. All patients fulfilled the Rome II criteria for IBS diagnosis (Thompson et al., 1999). By using standard, large-size, biopsy forceps, two sigmoid colon mucosal biopsy specimens, 10 cm apart, were collected from each participant using Affymetrix human U133 Plus 2.0 genechips (Aerssens et al., 2008). Presence/absence calls from negative probes were used to determine whether expression of a gene in a sample exceeded background expression (Warren et al., 2007). The microarray data as considered herein are log2(absolute expression) values which were corrected for a batch effect through an appropriate linear model. Details on the pre-processing which resulted in a fully regularised and normalised set of microarray gene expression readings are given in Aerssens et al. (2008). During this process, all personally identifiable information was stripped from the subjects. The dataset is available in both raw and filtered form from Aerssens (2007).
The second dataset is a set of microarray data for breast cancer patients, taken from Gorban and Zinovyev (2006). Affymetrix human U133a genechips were used to obtain data for 17,816 genes for each of 209 positive oestrogen receptor class (ER positive) and 77 negative oestrogen receptor class (ER negative) lymph node breast cancer patients. The resulting expressions were log2 transformed and normalised by scaling to a target probe set intensity. The process is fully described in Wang et al. (2005). Again, all personally identifiable information was stripped from the subjects.
Filtering techniques
It has been shown for some time that, in order to try to visualise a dataset of relatively high dimension, one can apply Principal Component Analysis (PCA) in order to identify directions of particular importance and high variance (see, for example, Wold et al. (1987)). However, PCA is not particularly useful as an initial step for processing microarray datasets since, as explained by Cumming and Wooff (2007), all the original variables are still required. It is for this reason that they discussed the idea of Principal Variables to reduce the dimension of the dataset instead. We intend to reduce the dimension of the datasets by explicit feature selection, that is, by selecting a few significant genes for which the expression values are particularly informative for classifying future cells into their corresponding diagnostic groups.
A common method of selecting significant genes is to rank each gene in order according to a common test statistic. We define the discriminatory ability of a gene as maximising some distance measure between the two groups' mean expression values for that gene. A common assumption for this type of classification is homoscedastic Gaussian group densities, as explained in the next section. Under this assumption, the null hypothesis states that the Mahalanobis distance, a standardised distance measure which places lower weightings on directions of higher variance, between the mean gene expression values of the two groups is equal to zero, and that the set of all these Mahalanobis distances, one for each gene, will be proportional to a common t-distribution. The proportionality constant will depend only on the classification group sizes, which will be the same for each gene tested as long as there is no missing data. Therefore, a greater Mahalanobis distance between the mean gene expression values of the two groups for a particular gene will result in a larger assigned corresponding t-test statistic, T, for that gene, and thus a lower p-value for the corresponding hypothesis test. A lower p-value implies that, under the assumption of the null hypothesis, the spread of gene expressions for that particular gene, with respect to the two groups, would be less likely to occur by chance. Therefore, a smaller p-value gives more evidence to reject the null hypothesis that a gene does not distinguish between the two groups.
The number of genes to select is a question of high importance. Usually a low budget of genes is preferential or necessary because of computational efficiency and the financial cost of processing a single gene expression. This question therefore leads the problem into the realms of decision-making, where the utility of the cost of finding the expression value of each additional gene has to be incorporated into a decision function alongside the potential benefits each additional gene may yield in terms of accurately predicting the correct group to which a future subject belongs.
Classification techniques
Once genes have been selected it is necessary to have a classifier which can take the expression values for the selected genes as inputs, and return a response indicating the presence or absence of the particular disease. There exist many such classifiers. Dudoit et al. (2002) explored many of these, including Fisher Linear Discriminant Analysis, Maximum Likelihood Discriminant rules, Nearest-Neighbour Classifiers and Classification trees, while looking to classify tumours using gene expression microarray data. Discriminant Analysis, and in particular Diagonal Linear Discriminant Analysis (DLDA), which we will be using, are reviewed below.
Discriminant analysis
Discriminant analysis is a statistical technique, first proposed by Fisher (1936, 1938), which allows the differences between two or more groups of objects to be studied with respect to several variables simultaneously (Klecka, 1980: 7–9), and thus define a function of these variables that attempts to distinguish between the groups. The idea is then to use this function to classify future unidentified individuals to exactly one group. We summarise the explanation of the Bayes' rule approach to discriminant analysis as given by McLachlan (1992: 4–10). Let πi be our prior probabilities that a sample belongs to group i. Let fi (x) be the group i probability density function. The group density functions are unlikely to be known and hence have to be estimated from the data using Bayes' rule.
Let Ω represent the whole input space. Let pij be the probability that an individual belonging to group j is misclassified as belonging to group i. In explicit terms, one has
where Ωi is the subspace of the whole input space that is classified into region i, and:
Let cij be the associated cost of misclassifying an individual belonging to group j to group i. For a particular classifier, Bayes' discriminant rule selects the group with minimum expected cost of misclassification. That is, for a future sample the selected group is
where G is the set of possible groups. Bayes' discriminant analysis aims to select that classifier which minimises the overall misclassification cost (McLachlan, 1992: 9). Having decided on this classifier, every combination of inputs now has a predicted group. We can split the input space up into sections for which the inputs lead to the same predicted group. The boundaries between these sections are called decision boundaries, as they are where we will change our decision about the predicted group of a point depending on which side of the boundary it is. A linear boundary is one which can be described by a linear function of the inputs, and a quadratic boundary is one which can be described by a quadratic function of the inputs.
Diagonal Linear Discriminant Analysis
Let us assume that each group population follows a multivariate normal distribution, which results in the decision boundaries being, at most, quadratic. If we assume homoscedasticity between the group populations as well, that is we assume all groups have a common covariance matrix:
it turns out that the decision boundaries are then linear (Hand, 1997: 31–32).
Diagonal discriminant analysis assumes that each group has a diagonal covariance matrix, implying independence between the parameters, or in our case, the genes. While often unrealistic, under this approach the number of parameters that need estimating for each group decreases considerably, namely, from order p2 to p.
Finally, DLDA involves making both these assumptions at once, that is, we assume we have a common diagonal covariance matrix. In the present article, we will use DLDA as our classifier for comparison purposes because of its speed and efficiency. As explained above, using more complicated forms of discriminant analysis requires estimating many more parameters and adds variance. Additional experiments have also shown that such techniques do not necessarily lead to an increased accuracy of classification for microarray experiments (Barker, 2011).
Validation
Once a classifier has been chosen, it is necessary to check the validity of the classifier to see how accurately the classifier may predict future subjects. If we assumed the accuracy of a classifier to be how well it predicted the response of the exact same data items used to build it, we run the risk that the model will very much have been fitted to perfectly predict the original data. This is especially a problem in high-dimensional situations, such as microarrays, when having p>>n variables makes fitting n observations trivial. This would not, however, help us to predict the group of further cases nor reflect the accuracy of doing so, and is known as overfitting.
In order to reduce the bias of the accuracy rate, we split the data into a training set and a test set. The training set is used to select significant genes and then build a classifier which only uses the gene expression values of these significant genes. The test set is then used to test the classifier by working out the proportion of the responses of data items in the test set which are correctly predicted by the classifier, which gives us an accuracy rate. The whole process is repeated many times so that lots of different combinations of training and test sets are considered, and the results are averaged to obtain an average accuracy rate. Throughout the present article a training set comprising of 75% of the data will be used for feature selection and classification, whilst the remaining 25% will be used to test the classifier to obtain a proportion of correctly predicted cases. Our experiments involved repeating this procedure for 3000 different randomly sampled training and test sets, and then averaging the resulting accuracies obtained over all of them.
All investigations carried out in this article, and the figures presented in it, were done in the computer program R-3-0 (R core team, 2013) with the inclusion of packages 'MASS' and 'supclust'. All R code is available from the first author, on request.
Correlation threshold
The main aim of the present article is to analyse the effect of the correlation threshold on the gene selection procedure and classification accuracies obtained. Application of the correlation threshold fits into the gene selection procedure as follows. We still place the genes in rank order, based on the value of the t-statistic for each one, and select a set of k genes from the list. However, instead of simply selecting the top k genes, each gene is considered in rank order, until we have selected k genes, and is selected only if it does not have a correlation higher than a certain threshold, b, with a gene of higher ranking. The motive behind the incorporation of a correlation threshold is to exclude genes from being selected if the information they provide is similar to that given by a gene that has already been selected. A gene will be less likely to provide additional classification information if it has a high correlation with a gene already selected.
Application of methods to the IBS dataset
The results of repeatedly constructing a DLDA classifier using the top k genes from the t-statistic rank list, for the IBS microarray data, for each of 3000 different combinations of training and test sets, for five different values of k, are shown in the top line of Table 1. We can see that increasing the number of genes within this small range only improves the accuracy a little, and in fact, although the differences are not huge, k=30 seems to work a little better than k=40 in this example. The second line of Table 1 shows the results of the same classification procedure, but using a correlation threshold of b=0.8. Although it may be observed that the individual change of correlation threshold from b=1, that is no correlation threshold, to b=0.8 seems to have slightly lowered the accuracies obtained from the classifier, it is worth noting that the local maximum for b=0.8 is obtained for a smaller number of genes as compared to b=1. A possible explanation is that we may expect correlated genes to be representing the same (or similar) biological pathways, so that removing some correlated genes may result in us requiring less genes to represent many pathways.
| K | 10 | 15 | 20 | 30 | 40 |
|---|---|---|---|---|---|
| b=1 prediction accuracy (%) | 69.05 | 69.86 | 70.32 | 70.04 | 68.71 |
| b=0.8 prediction accuracy (%) | 68.36 | 68.90 | 68.78 | 68.41 | 68.58 |
Table 1: A comparison of the average prediction accuracies achieved when selecting different numbers of genes, k, to include in the application of a DLDA classifier on the IBS dataset, with correlation threshold values of b=1, that is, no correlation threshold, and b=0.8.
The results of varying the correlation threshold, b, and number of genes, k, on the DLDA average classification accuracy of the IBS dataset, are presented in Figure 2. We can observe that, in this example, using b=1, that is, in effect no correlation threshold, seems to give the most accurate classifications. However, it is once again noted that lowering the correlation threshold seems to move the maximum of the accuracy curves towards the left.
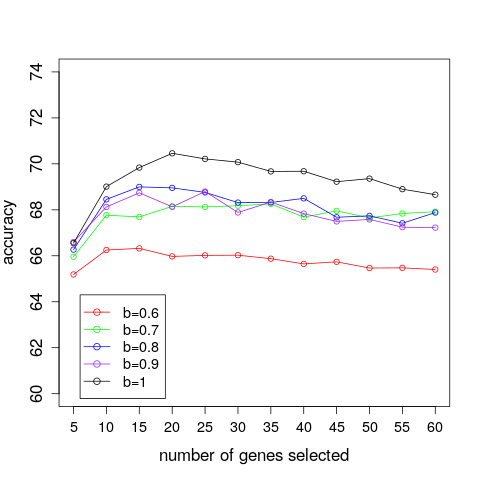
Figure 2: A comparison of the average prediction accuracies achieved for different numbers of selected genes and correlation threshold values when applying DLDA on the IBS dataset (the displayed accuracies are average values over the accuracies obtained from the 3000 test sets).
Given these results it would be interesting to know how many genes are being removed from the original list ranking by different correlation threshold values. In Table 2, we present the number of genes removed from the full ranking when k=10 and k=20, for each correlation threshold, b, of 0.6, 0.7, 0.8, 0.9 and 1. This analysis has been done using the full sample dataset. We can see that the number of genes removed dramatically increases for b=0.6.
| Correlation threshold, b | 1 | 0.9 | 0.8 | 0.7 | 0.6 |
|---|---|---|---|---|---|
| Genes removed for k=10 | 0 | 3 | 3 | 8 | 17 |
| Genes removed for k=20 | 0 | 6 | 6 | 15 | 66 |
Table 2: A comparison of the number of genes removed from the full gene ranking of the IBS dataset for different correlation thresholds and numbers of genes.
The Affymetrix identifiers of the top genes that we obtained by using these methods on the whole IBS dataset were as follows: 201762_s_at, 211368_s_at, 1552701_a_at, 211367_s_at, 211366_x_at, 225809_at, 224523_s_at, 229369_at, 1552703_s_at, 222233_s_at, 242826_at, 203696_s_at, 204687_at, 226622_at, 239376_at, 212034_s_at, 223655_at. Many of these genes are the same as those found in Aerssens et al. (2008).
Application of methods to the breast cancer dataset
We present, in Figure 3, the results of varying the correlation threshold, b, and number of genes, k, on the DLDA average classification accuracy of the breast cancer dataset. We can see that for smaller numbers of genes a suitable correlation threshold seems to have a positive effect, since lower correlation thresholds seem to yield slightly higher accuracies. As the number of genes increases, it would appear that a correlation threshold is not beneficial since a threshold value of b=0.6 yields lower accuracies than the other correlation threshold values. These qualitative statements can be supported by considering the standard errors of the accuracies, for which we refer to Einbeck et al. (2015). We additionally present, in Figure 4, accuracy curves for the IBS and the breast cancer data within one plot. One observes that any differences in prediction accuracy which can be achieved by either changing the number of included genes or the correlation threshold are rather marginal when compared to differences in prediction errors between different datasets.
The Affymetrix identifiers of the top genes obtained by using these methods on the whole breast cancer dataset are as follows: 205225_at, 209603_at, 209604_s_at, 212956_at, 215867_x_at, 209173_at, 209602_s_at, 214164_x_at, 204508_s_at, 203963_at, 205186_at, 202088_at, 203929_s_at, 215304_at, 200670_at, 218976_at, 205009_at, 218195_at, 212195_at, 218211_s_at.
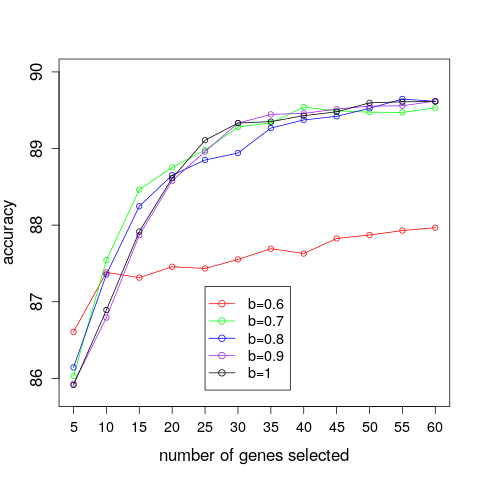
Figure 3: A comparison of the average prediction accuracies achieved for different numbers of selected genes and correlation threshold values when applying a DLDA classifier on the breast cancer dataset.
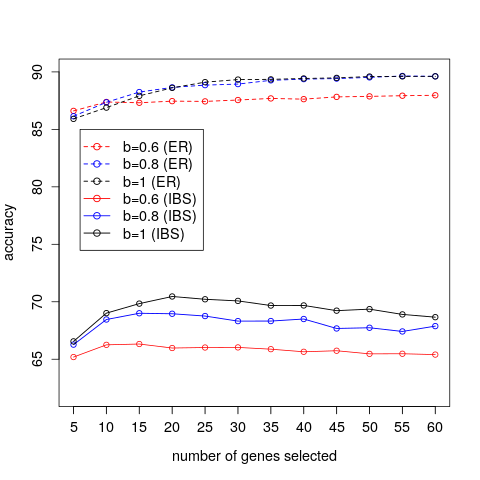
Figure 4: A comparison of the average prediction accuracies achieved for the IBS and the breast cancer (ER) data.
Conclusion and further research
We have tested the idea of applying a correlation threshold to the feature selection process of microarray data classification on two microarray datasets. For the IBS data, it seemed that the effect of including a correlation threshold was to decrease the accuracy of the classifier. This may tend to suggest that, although incorporating a correlation threshold did result in some correlated genes being removed, doing so was not beneficial. For the breast cancer data, the correlation threshold had a much more positive effect when the number of genes required was small. These results together suggest that the most beneficial correlation threshold is dependent on the particular microarray data analysis, and that applying a correlation threshold at all may only be worth considering if the number of genes required is restrictively small.
It is perhaps not that surprising that the correlation threshold may only be beneficial for smaller numbers of genes, since it is then that filling up one of the few available genes with a possibly redundant gene will be much more costly. The results seem to show, however, that even though highly correlated genes have been removed in both cases, the differences in the resulting accuracies are not particularly large, even though in some cases significant.
For a particular microarray analysis experiment, it may be that the optimal correlation threshold, b, needs estimating from the data. This may be a relatively straightforward task for a fixed number of genes, k. However, if k also needs optimising with respect to accuracy, inclusive of a decision-theoretic penalty for increasing the number of genes, then the optimisation procedure may become rather complex.
Previous research has looked into similar concepts. However, in general, the accuracy curves presented in this article are smoother and have better identifiable maxima compared to those in the literature (Jaeger et al., 2003). Jaeger et al. (2003) mention the idea of a threshold which removed genes from the ranking list if they correlated with a gene already selected. The difference with the correlation threshold considered within the present article is that genes are removed if they correlate with a gene of lower p-value, or higher test statistic, regardless of whether the gene was selected itself or not. As an example, suppose that we have genes A, B and C ranked in alphabetical order. Mathematically speaking, if gene C has a high correlation with gene B, and gene B has a high correlation with gene A, it does not necessarily follow that gene C has such a high correlation with gene A. However, under the method proposed herein, gene C would still not be selected since it had a high correlation with gene B, regardless of the fact that gene B was not actually chosen itself. By the nature of the biological application of microarray data, this is unlikely for the genes which are highly ranked in the ranking order due to the spread of expression values between the two groups which must occur for this to happen. However, even if such a case should occur, it is possible that, by the connection of correlations to an intermediary gene, the two genes are still from the same pathway, and thus maybe both should still not be chosen.
A further option, which is considered in some pathway analysis techniques (see for example, Jaeger et al. (2003)), is to include only the most intermediary gene, for example, in the scenario above, gene B, which correlates with both of the others, assuming this to be most representative of that particular pathway. Yeung and Bumgarner (2003) considered a similar notion of a correlation threshold in connection with a shrinkage threshold for removing genes to increase the feature stability.
Alternatives to selecting only the most significant individual genes have also been considered in past papers, including Hotelling's two-sample T²-statistic for groups of multiple variables (see, for example, Xiong et al. (2002)). It is not possible, due to computational limitations or efficiency, to test every possible collection of genes in this way and obtain a test statistic for each one. However, the multiple variable test statistic can be useful when it is necessary to decide between several sets of high-ranking genes. An alternative approach for a problem involving just two classification groups is the 'Top Scoring Pair' approach, as used by Edelman et al. (2009) and Zhao et al. (2010), which considers the relative difference of gene expression values between all possible pairs of genes so as to pick various pairs of genes for which the relative difference between their two gene expression values is predictive of correct group classification. This idea has recently (Yang and Naiman, 2014) been extended to top scoring sets of k genes for k-class problems. It is hoped that the ideas considered in this article for removing highly correlated individual genes may be extended to these more recent methodologies in order to eliminate highly correlated and redundant pairs or sets of genes from the building of a classifier.
In conclusion, we have shown that very good classification rates can be achieved in certain applications through the use of some standard methods of gene selection. We have also seen that the application of a correlation threshold will often lead to a decrease of the classification accuracy. However, when the required number of genes is restrictively small, (which may be the case due to financial or computational reasons), we did observe situations in which the application of a threshold b<1 turned out to be beneficial.
Acknowledgements
The principal author's research was supported by an Engineering and Physical Sciences Research Council (EPSRC) vacation bursary, as well as a small grant given by The Institute of Mathematics and its Applications (IMA). The authors wish to thank Janssen Pharmaceutica N. V., Beerse, Belgium, for providing the IBS data.
List of figures
Figure 1: Comparing the plots of the expression values for a potentially informative gene and an uninformative gene, both taken from a microarray analysis dataset for IBS.
Figure 2: A comparison of the average prediction accuracies achieved for different numbers of selected genes and correlation threshold values when applying DLDA on the IBS dataset (the displayed accuracies are average values over the accuracies obtained from the 3000 combinations of training and test sets).
Figure 3: A comparison of the average prediction accuracies achieved for different numbers of selected genes and correlation threshold values when applying a DLDA classifier on the breast cancer dataset.
Figure 4: A comparison of the average prediction accuracies achieved for the IBS and the breast cancer (ER) data.
List of tables
Table 1: A comparison of the average prediction accuracies achieved when selecting different numbers of genes, k, to include in the application of a DLDA classifier on the IBS dataset, with correlation threshold values of b=1, that is, no correlation threshold, and b=0.8.
Table 2: A comparison of the number of genes removed from the full gene ranking of the IBS dataset for different correlation thresholds and numbers of genes.
References
Aerssens, J. (2007), 'E-TABM-176 - Transcription profiling of sigmoid colon mucosal biopsies from irritable bowel syndrome patients and healthy control subjects', Array Express, available at http://www.ebi.ac.uk/arrayexpress/experiments/E-TABM-176/
Aerssens, J., M. Camilleri, W. Talloen, L. Thielemans, H. W. H. Gohlmann, I. Van den Wyngaert, T. Thielemans, R. De Hoogt, C. N. Andrews, A. E. Bharucha, P. J. Carlson, I. Busciglio, D. D. Burton, T. Smyrk, R. Urrutia and B. Coulie (2008), 'Alterations in mucosal immunity identified in the colon of patients with Irritable Bowel Syndrome', Clinical Gastroenterology and Hepatology, 6, 194–205
Barker, K. (2011), 'Microarray feature extraction', 4H Master of Mathematics project report, Durham University
Chen, D., Z. Liu, X. Ma and D. Hua (2005), 'Selecting genes by test statistics', Journal of Biomedicine and Biotechnology, 2, 132–38
Cumming, J. A. and D. A. Wooff (2007), 'Dimension reduction via principal variables', Computational Statistics and Data Analysis, 52, 550–65
Dudoit, S., J. Fridlyand and T. P. Speed (2002), 'Comparison of discrimination methods for the classification of tumours using gene expression data', Journal of the American Statistical Association, 97 (457) ), 77-87
Edelman, L. B., G. Toia, D. Geman, W. Zhang and N. D. Price (2009), 'Two-transcript gene expression classifiers in the diagnosis and prognosis of human diseases', BMC Genomics, 10 (583), available at http://dx.doi.org/10.1186/1471-2164-10-583
Einbeck, J., S. E. Jackson and A. Kasim (2015), 'A summer with genes: Simple disease classification from microarray data', Mathematics Today, 51, 186–88
Fisher, R. A. (1936), 'The use of multiple measurements in taxonomic problems', Annals of Eugenics, 7 (2), 179–88
Fisher, R. A. (1938), 'The statistical utilisation of multiple measurements', Annals of Eugenics, 8 (4), 376–86
Golub, T. R., D. K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J. P. Mesirov, H. Coller, M. L. Loh, J. R. Downing, M. A. Caligiuri, C. D. Bloomfield and E. S. Lander (1999), 'Molecular classification of cancer: class discovery and class prediction by gene expression monitoring', Science, 286, 531–37
Gorban, A. and A. Zinovyev (2006), Workshop 'Principal Manifolds – 2006', August 24–26, 2006, Leicester, UK, available at http://www.ihes.fr/~zinovyev/princmanif2006/, Dataset I
Hand, D. J. (1997), Construction and Assessment of Classification Rules, Chichester: Wiley
Jaeger, J., R. Sengupta and W. L. Ruzzo (2003), 'Improved gene selection for classification of microarrays', Proceedings from Pacific Symposium on Biocomputing (PSB) conference, 2003, Lihue, Hawaii, USA, 8, 53–64
Klecka, W. R. (1980), Discriminant Analysis, London: Sage Publications
McLachlan, G. J. (1992), Discriminant Analysis and Statistical Pattern Recognition, New York: Wiley
National Human Genome Research Institute (2016), 'Biological Pathways', available at https://www.genome.gov/27530687/biological-pathways-fact-sheet/
Oti, M. and H. G. Brunner (2007), 'The modular nature of genetic diseases', Clinical Genetics, 71 (1), 1–11
Quackenbush, J. (2006), 'Microarray analysis and tumor classification', New England Journal of Medicine, 354, 2463–72
R core team (2013), 'R: A Language and Environment for Statistical Computing', R Foundation for Statistical Computing, Vienna, Austria, http://www.R-project.org
Thompson, W. G., G. F. Longstreth, D. A. Drossman, K. W. Heaton, E. J. Irvine and S. A. Müller-Lissner (1999), 'Functional bowel disorders and functional abdominal pain', Gut, 45 (Supp. 2), 43–47
Tusher, V. G., R. Tibshiriani and G. Chu (2001), 'Significance analysis of microarrays applied to the ionising radiation response', Proceedings of the National Academy of Sciences, 98, 5116–21
van't Veer, L. J., H. Dai, M. J. van de Vijver, Y. D. He, A. A. Hart, M. Mao, H. L. Peterse, K. van der Kooy, M. J. Marton, A. T. Witteveen, G. J. Schreiber, R. M. Kerkhoven, C. Roberts, P. S. Linsley, R. Bernards and S. H. Friend (2002), 'Gene expression profiling predicts clinical outcome of breast cancer', Nature, 415 (6871), 530–36
Wang, Y., J. G. Klijn, Y. Zhang, A. M. Sieuwerts, M. P. Look, F. Yang, D. Talantov, M. Timmermans, M. E. Meijer-van Gelder, J. Yu, T. Jatkoe, E. M. J. J. Berns, D. Atkins and J. A. Foekens (2005), 'Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer', The Lancet, 365 (9460), 671–79
Warren, P., D. Taylor, P. G. V. Martini, J. Jackson and J. Bienkowska (2007), 'PANP – a new method of gene detection on oligonucleotide expression arrays', IEEE 7th International Symposium on Bioinformatics and Bioengineering, 108–15
Wold, S., K. Esbensen and P. Geladi (1987), 'Principal Component Analysis', Chemometrics and Intelligent Laboratory Systems, 2, 37–52
Xiong, M., J. Zhao and E. Boerwinkle (2002), 'Generalised T² test for genome association studies', The American Journal of Human Genetics, 70 (5), 1257–68
Yang, S. and D. Q. Naiman (2014), 'Multiclass cancer classification based on gene expression comparison', Statistical Applications in Genetics and Molecular Biology, 13 (4), 477–96
Yeung, K. Y. and R. E. Bumgarner (2003), 'Multiclass classification of microarray data with repeated measurements: application to cancer', Genome Biology, 4 (12), available at http://genomebiology.com/2003/4/12/r83
Zhao, H., C. J. Logothetis and I. P. Gorlov (2010), 'Usefulness of the top-scoring pairs of genes for prediction of prostate cancer progression', Prostate Cancer and Prostatic Diseases, 13 (3), 252–59
To cite this paper please use the following details: Jackson, S.E., Einbeck, J., Kasim, A. and Talloen, W. (2016), 'The Correlation Threshold as a Strategy for Gene Filtering, with Application to Irritable Bowel Syndrome and Breast Cancer Microarray Data', Reinvention: an International Journal of Undergraduate Research, Volume 9, Issue 2, http://www.warwick.ac.uk/reinventionjournal/archive/volume9issue2/jackson Date accessed [insert date]. If you cite this article or use it in any teaching or other related activities please let us know by e-mailing us at Reinventionjournal at warwick dot ac dot uk.