
The Gas Phase
Project Overview
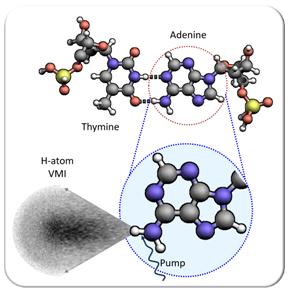 Processes which involve the absorption of light play an integral role in our day-to-day lives. Nature has carefully chosen our molecular building blocks so that potentially devastating effects of ultraviolet (UV) radiation are by-passed. Some of the most important molecular building blocks, the DNA bases (adenine, thymine, guanine and cytosine), absorb ultraviolet radiation very readily. However, once absorbed, this energy is efficiently diffused through harmless molecular relaxation pathways which reduce the risk of molecular breakdown and therefore photochemical damage.
Processes which involve the absorption of light play an integral role in our day-to-day lives. Nature has carefully chosen our molecular building blocks so that potentially devastating effects of ultraviolet (UV) radiation are by-passed. Some of the most important molecular building blocks, the DNA bases (adenine, thymine, guanine and cytosine), absorb ultraviolet radiation very readily. However, once absorbed, this energy is efficiently diffused through harmless molecular relaxation pathways which reduce the risk of molecular breakdown and therefore photochemical damage.
The timescales of the photoresistive pathways must be very fast for them to compete effectively with the detrimental paths. It is becoming increasingly clear however that, although ultrafast measurements with lasers reveal very fast relaxation pathways [1,2], more refined experiments are required to test the ever increasingly sophisticated calculations that model the theory behind these pathways. One of the key pathways implicated, first by theory [3] and subsequently by numerous experiments [4], involves relaxation via repuslive 1πσ* states localised along X-H (X = O or N) bond coordinates. When excess photon energy is coupled onto these states this may result in either rapid repopulation of a vibrationally hot electronic ground state via effective conical intersections or ultrafast X-H fission and the production of high kinetic energy H-atoms.
Our research aims to identify these pathways and completely characterize them by studying the dynamics of these systems in isolated environments such as molecular beams. By combining molecular beam methodologies with time-resolved velocity map ion imaging (TR-VMI), we will begin to understand, not only the photoresistive mechanisms of the individual bases, but the more realistic scenario, the base-pair. We hope to transfer the knowledge gained from these measurements to study these processes in the liquid phase with the aid of transient absorption measurements, mimicking conditions in human cells.
To elucidate and classify these pathways as effectively as possible we also have collaborations with a number of other research groups:
- Prof. Mike Ashfold (Bristol University, UK):H Rydberg atom photofragment translational spectroscopy.
- Prof. Martin Paterson (Heriot-Watt University, UK): High level ab initio calculations, typically complete active space (CAS) methodolgies.
- Dr Dave Townsend (Heriot-Watt University, UK):Soft molecular desorption, molecular dynamics of biomolecules and tuneable VUV light generation.
- Dr Susanne Ullrich (University of Georgia (GA), USA): Time-resolved photoelectron/photoion coincidence measurements and time-resolved magnetic bottle photoelectron spectroscopy.
- Dr Jan Verlet (University of Durham, UK): Time-resolved photoelectron imaging of biomolecule ions.
- Prof. Tim Zwier (Purdue University, USA): High-resolution spectroscopy and molecular dynamics.
References:
[1] C.E. Crespo-Hernandez et al., Chem. Rev., 104 (2004) 1977
[2] C.Z. Bisgaard et al., ChemPhysChem, 10 (2009) 101
[3] A.L. Sobolewski et al., Phys. Chem. Chem. Phys., 4 (2002) 1093
[4] M.N.R Ashfold et al., Science, 312 (2006) 1637
Experiment Details
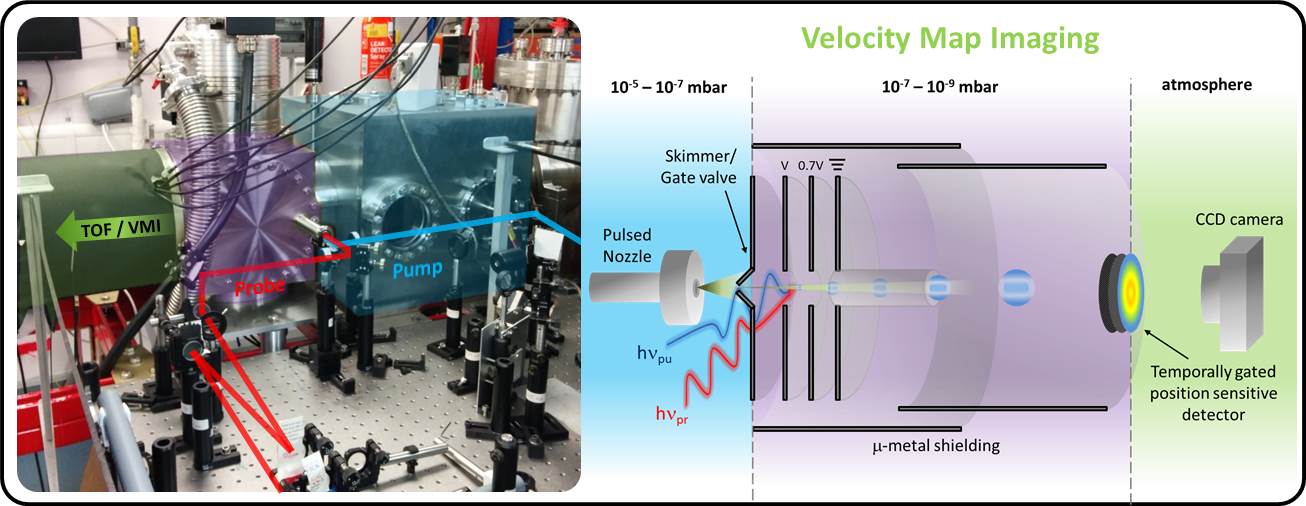
Biomolecules are studied in a differentially pumped molecular beam machine (above, left) consisting of source (blue) and interaction chambers (green). The source houses an Even-Lavie pulsed valve [1] while the interaction region contains a velocity map imaging (VMI) spectrometer [2]. Pump and probe laser pulses are derived from a commercial femtosecond laser system (Spectra-physics, Tsunami oscillator and Spitfire regenerative amplifier) which generates 800 nm pulses (~40 fs) in a kHz pulse train. These fundamental pulses are then converted using optical parametric amplification (Topas) and harmonic generation techniques to provide both pump and probe. Pump and probe are temporally delayed with respect to each other and perpendicularly intersect the seeded molecular beam pulse. The Pump induced X-H fission yields H-atoms which are ionised using a 2 + 1 REMPI scheme (2s ← 1s). The energies and angular distributions of the H+ ions are then mapped onto a time gated position sensitive detector (MCP + phosphor screen) placed at the terminus of the VMI drift tube (see above, right). Light emitted from the screen is subsequently captured using a CCD camera, producing a H+ image. Images are finally reconstructed using a polar onion-peeling algorithm [3] to generate the associated H-atom total kinetic energy release (TKER) spectra at each temporal delay.
References:
[1] U. Even et al., J. Phys. Chem., 112 (2000) 8068
[2] A. Eppink and D. Parker, Rev. Sci. Instrum., 68 (1997) 3477
[3] G.M. Roberts et al., Rev. Sci. Instrum., 80 (2009) 053104
Example Studies
1. Tyrosine and Related Derivatives
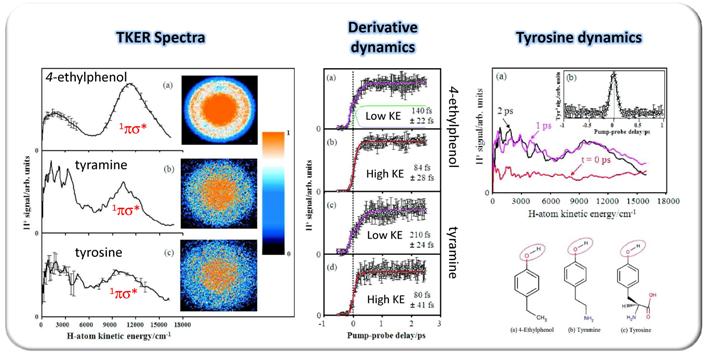
H-atom elimination studies on the amino acid tyrosine have been conducted which implicate the active participation of 1πσ* states following photoexcitation at 200 nm (results displayed below) [1]. For comparison we have also investigated the dynamics occuring in the related species 4-ethylphenol and tyramine, in an attempt to understand the evolution of the O-H fission dynamics from tyrosine's UV chromophore phenol, which we have also reported upon [2,3], to the amino acid itself. All three species are shown on the bottom right.
The results show below for 4-ethylphenol clearly shows that ultrafast H-atom elimination occurs following dissociation of the O-H bond after excitation at 200 nm (fast KE component in TKER spectrum). The dynamics of the H+ transient in tyramine and the KE features of the H-atom spectra in tyrosine coupled with the parent tyrosine transient data (not shown) also strongly suggests that, following excitation at 200 nm, H-atom elimination occurs through the same coordinate, i.e., the phenol-like O-H bond as exhibited by the chromophore phenol. This provides direct evidence of the active participation of 1πσ* states from the subunit of the amino acid tyrosine (4-ethylphenol) to tyrosine itself.
References:
[1] A. Iqbal and V.G. Stavros, J Phys. Chem. Lett., 1 (2010) 2274
[2] A. Iqbal et al., J Phys. Chem. A, 113 (2009) 8157
[3] A. Iqbal et al., J Phys. Chem. A, 112 (2008) 9531
2. Quantum Beating in Phenol, Guaiacol and Syringol
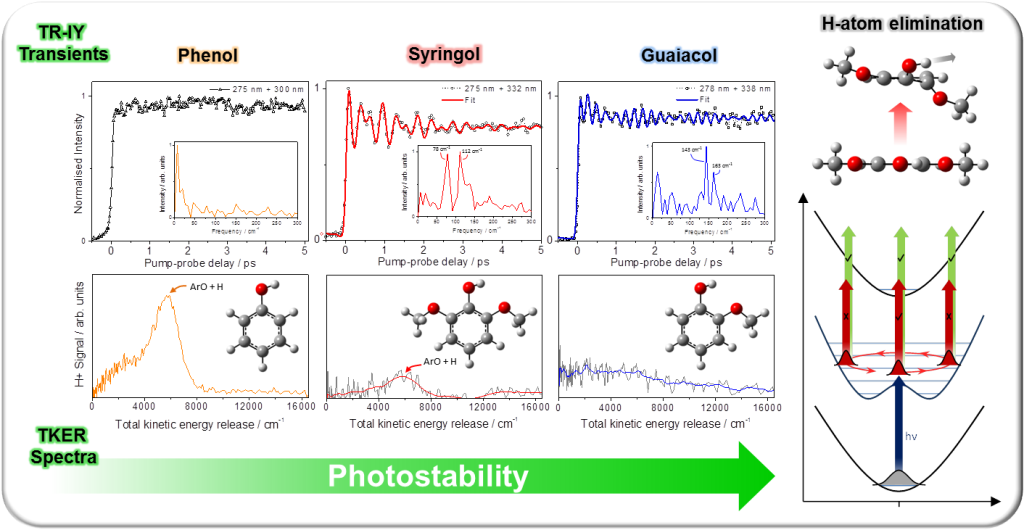
Following on from our investigations into phenols and substituted derivatives, the photoinduced dynamics of the lignin building-blocks syringol, guaiacol and phenol (structures shown below) were studied using time-resolved ultrafast femtosecond spectroscopy. Following irradiation of syringol and guaiacol with ultraviolet light, the excited state dynamics display a quantum beat pattern (see TR-IY transients below) attributed to out-of-plane OH torsion and methoxy (OMe) torsion/flapping motions.[1] These motions arise due to a dramatic planar → non-planar geometry change folowing excitation, which is absent in phenol. An example of this dramatic distortion is shown for syringol in the top-right corner of the figure below.
Interestingly, for syringol, the geometry change is pronounced enough such that the degree of intramolecular H-bonding (between OH and OMe groups) is reduced, enabling H-atom elimination from the OH group. For guaiacol, H-bonding is preserved after excitation, despite the geometry change, and prevents O-H bond fission.[2] This behavior affects the propensities for forming undesired phenoxyl (ArO) radical sites in these three lignin chromophores, and provides important insight into their relative ‘photostabilities’ within the larger biopolymer, lignin.
References:
[1] Dean, J. C.; Navotnaya, P.; Parobek, A. P.; Clayton, R. M.; Zwier, T. S. J. Chem. Phys. 139 (2013) 144313
[2] Chatterley, A. S.; Young, J. D.; Townsend, D.; Zurek, J. M.; Paterson, M. J.; Roberts, G. M.; Stavros, V. G., Phys. Chem. Chem. Phys. 15 (2013) 6879
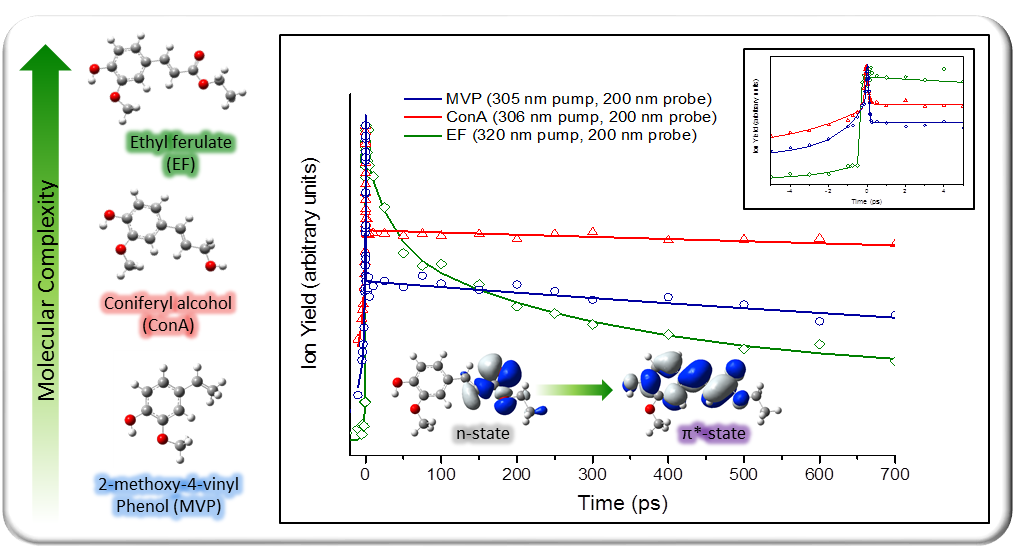
3. Sunscreens and Related Derivatives
Despite the widespread use of sunscreens, very little is known regarding the photochemistry of the molecules used in these lotions, designed to protect human skin from the harmful effects of the UV radiation from the Sun. The photochemistry of large biomolecules such as those present in the composition of sunscreen lotions may result, for example, in the formation of free radicals responsible for DNA damage, and, consequently, cancer [1]. Studying these compounds is essential to evaluating their safety and provides useful information for the development of more efficient sunscreens.
Theoretical work by Karsili et al. [2] suggests OH/OCH3 bond extensions, ring centred out-of-plane deformations and E/Z photoisomerism as possible internal conversion pathways for ferulic acid, a common sunscreen component. The ππ* and nπ* states of ferulic acid are found to be close in energy and possess multiple conical intersections, suggesting possible deactivation pathways exist between these states.
Ferulic acid could not be successfully vaporised in our system as it fragments under heat. Ethyl ferulate, the ethyl ester of ferulic acid was studied instead, since a similar behaviour is expected. Preliminary studies so far support the deactivation of photoexcited ethyl ferualte via an nπ* state.
References:
[1] Maslin, D. L., International Journal of Dermatology 53, (2014) 1319.
[2] Karsili, T. N. V. et al., J. Phys. Chem. A 118, (2014) 11999.