
A Guide to Gibson Assembly Design
Overview
Gibson Assembly is an extremely useful DNA assembly method developed by Daniel Gibson at the J. Craig Venter Institute. I use it in place of standard restriction enzyme based molecular cloning to create circular DNA plasmids for use E. coli and S. cerevisiae. The basic premise is shown in the diagram to the right and is as follows: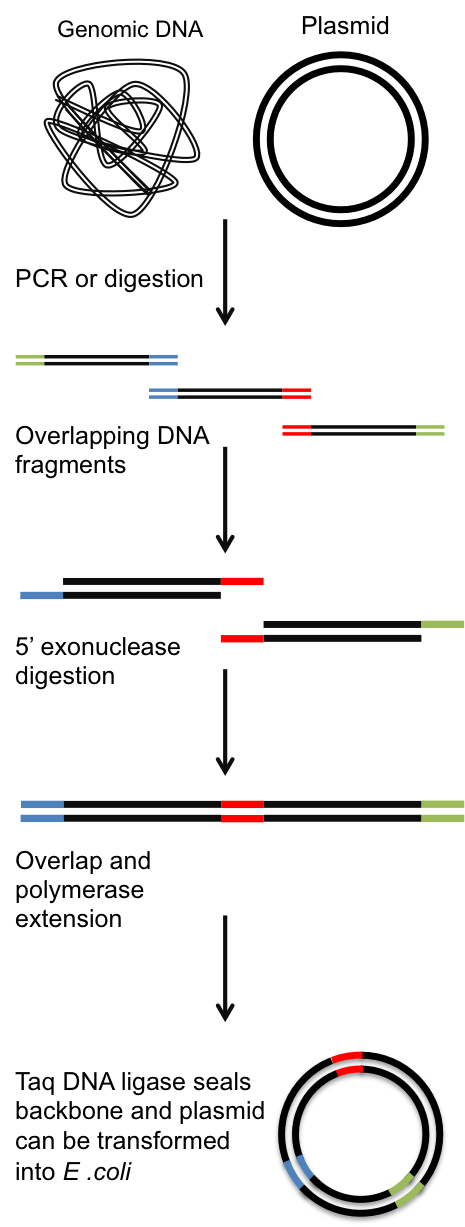
- Generation of DNA fragments with overlapping ends - either by restriction digest or PCR.
- T5 5' exonuclease digestion of DNA fragments to yield 'sticky' ends.
- Complementary base pairing of overlapping ends allows fragments to form circular plasmid.
- Phusion DNA polymerase fills in gaps in the plasmid.
- Taq Ligase seals the nicks in the DNA backbone.
Due to the ability to precisely define overlaps in oligonucleotide primers, Gibson assembly becomes a seamless process, in that no scar is present in the plasmid. Primers are easy to design and available commercially, and so Gibson assembly allows any substrate that is accessible to PCR to be incorporated into new DNA elements, this include genomic DNA, plasmids and artificial chromosomes. Since overlaps can be introduced in a single primer, plasmid backbones can also be digested with restriction enzymes and PCR fragments introduced via Gibson. This methods has an added advantage with enzymes leaving a 5’ overhang, in that they are digested by the 5’ exonuclease, removing the restriction site scar (see below). Here I will outline how I design my Gibson assemblies to give the perfect plasmid.
The first step in any molecular cloning process is to define what you want to build. I generally build plasmids for yeast and bacteria using commercial or openly available plasmid backbones from Addgene. In this example we will work through the design of a Gibson assembly to insert 4 DNA fragments into a plasmid backbone, to yield a usable yeast centromeric plasmid.
Step 1 – Plasmid Design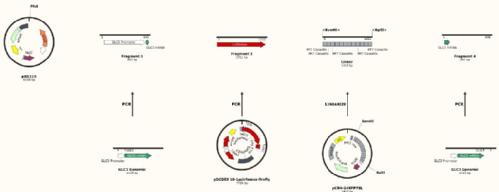
The best way to design your desired plasmid is with a DNA manipulation software package. There are many of these available for free and commercially. In our lab we use SnapGene, which is a user-friendly system with a number of simulation tools, including one for Gibson assembly, that allow easy planning of molecular cloning procedure.
First, define the exact DNA sequences that you wish to assemble in the reaction. DNA sequences for ORFs and non-coding regions can be found in online repositories, for example the Saccharomyces genome database has sequences for all S. Cerevisiae ORFs, and other databases contain promoter sequences and 5' and 3' mRNA UTRs.  Once you know the sequences you want to join and that you can access them in the lab (e.g. have the correct plasmids or cell lines) you can arrange them in the order you want in your manipulation software. To the right you can see the 4 sequences I have chosen from various sources, as well as the plasmid backbone, and how I will be isolating them in the lab. I have then Copy/Pasted them into the digested backbone plasmid sequence in the order I wanted them, and circularised by joining the 2 ends to get the desired plasmid sequence, shown to the left. Remember when using restriction cloning than you must remove any 5' overhangs that are generated before compiling your plasmid map, as they will be degraded by the 5' exounclease during the reaction. Once you have generated your plasmid map from your fragments, you can move on to designing the oligonucleotide primers to generate the overlapping ends. You can see from my fragments than I am using restriction enzymes to isolate fragment 3, this fragment contains stem-loop structures that make it difficult to PCR. To allow me to use the gibson reaction to introduce this fragment i therefore need to include longer overlaps on fragments 2 and 4, to compensate for the lack of overlap on fragment 3, see below.
Once you know the sequences you want to join and that you can access them in the lab (e.g. have the correct plasmids or cell lines) you can arrange them in the order you want in your manipulation software. To the right you can see the 4 sequences I have chosen from various sources, as well as the plasmid backbone, and how I will be isolating them in the lab. I have then Copy/Pasted them into the digested backbone plasmid sequence in the order I wanted them, and circularised by joining the 2 ends to get the desired plasmid sequence, shown to the left. Remember when using restriction cloning than you must remove any 5' overhangs that are generated before compiling your plasmid map, as they will be degraded by the 5' exounclease during the reaction. Once you have generated your plasmid map from your fragments, you can move on to designing the oligonucleotide primers to generate the overlapping ends. You can see from my fragments than I am using restriction enzymes to isolate fragment 3, this fragment contains stem-loop structures that make it difficult to PCR. To allow me to use the gibson reaction to introduce this fragment i therefore need to include longer overlaps on fragments 2 and 4, to compensate for the lack of overlap on fragment 3, see below.
Step 2 - Oligonucleotide Design
During any Gibson assembly reaction, one of two DNA fragment types will be joined, either a PCR of a restriction digest fragment. The design of primers to generate overlaps varies depending on which fragments are being joined. Remember that at each joint in your plasmid, at least one side much be a PCR fragment to allow for the introduction of these overlaps. Below I will outline how to design primers for joining either 2 PCR fragments, or a PCR fragment to a restriction fragment. Ideally you want your primer to have a binding region with a Tm of around 60oC and for the overlap to have as high a Tm as possible to ensure tight binding during the gibson reaction. As with all primer design you should use software (available freely online) to check for secondary structures and dimerization in your primer pairs. You should also remember that most oligonucleotide synthesis companies have different prices depending on the length of your sequence, so try to keep your primers short enough to fall into the lower price bands, for example 60 bp or below for IdT.
PCR fragment to PCR fragment
We will start wth joining 2 PCR fragments as these primers are the easiest to design. In your plasmid map, find the region where your 2 fragments meet. Starting with either fragment, select a region of sequence starting from the joint that gives a Tm of around 60oC as below, make sure to include a G/C anchor at the 5' end of the primer. This is now the binding region of your primer. Repeat this process with the other fragment to find a binding region with the correct Tm, as shown below.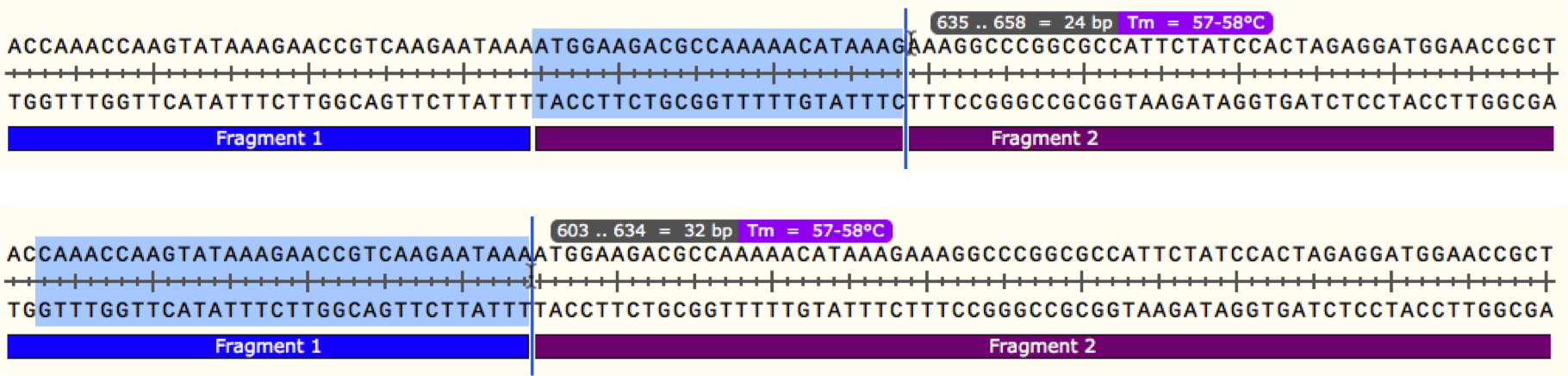 Once you have the binding regions for your primers, you next need to add the overlapping regions. In general, an overlap of 40 bp yields a sufficient Tm for the gibson reaction, so if we extend each of our primers from the 5' end by 20 bp, we will have 40 bp of overlap, and can measure the Tm of that region, as below.
Once you have the binding regions for your primers, you next need to add the overlapping regions. In general, an overlap of 40 bp yields a sufficient Tm for the gibson reaction, so if we extend each of our primers from the 5' end by 20 bp, we will have 40 bp of overlap, and can measure the Tm of that region, as below.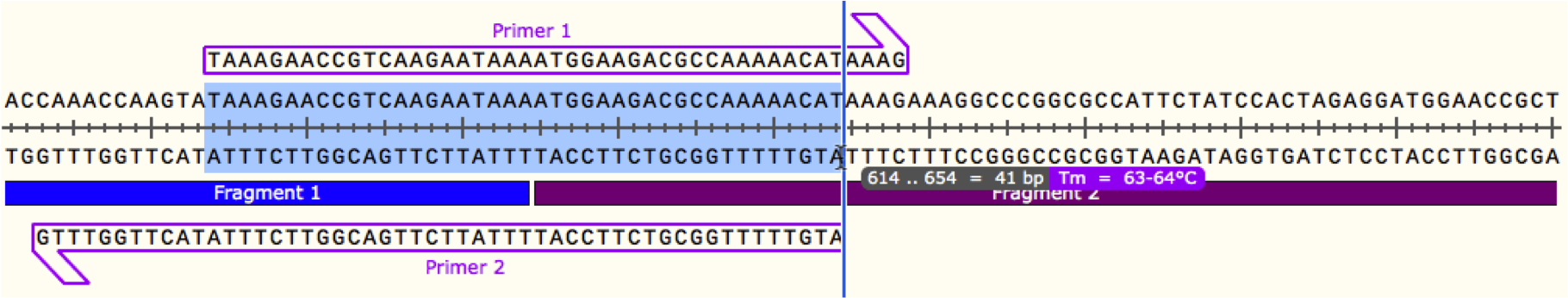 Now we have primer sequences for both sides of the joint, with sufficient binding to both the templates and each other to allow the gibson assembly reaction to proceed, as well as being small enough for the lower price bracket for synthesis. If you are just using PCR fragments you can repeat this process for each joint, and then simply amplify each fragment and assemble. If you are using some restriction fragments or backbone, you will need to design slightly different primers to compensate for the lack of overlap.
Now we have primer sequences for both sides of the joint, with sufficient binding to both the templates and each other to allow the gibson assembly reaction to proceed, as well as being small enough for the lower price bracket for synthesis. If you are just using PCR fragments you can repeat this process for each joint, and then simply amplify each fragment and assemble. If you are using some restriction fragments or backbone, you will need to design slightly different primers to compensate for the lack of overlap.
PCR fragment to restriction fragment
The main difference with joining a PCR fragment to a restriction fragments is that the restriction fragment cannot have any extra overlap introduced into it without additional modifications. To compensate for this we need to make the tail of the PCR fragment primer longer, so that the overlap is still sufficient for the reaction. We also need to consider what form of overlap the restriction enzyme that you are using generates. If this overlap is 5' then it will be degraded during the reaction so it can be excluded from your design, but if it is 3' then it must be included as it cannot be degraded. This should be handled at the level of plasmid design as mentioned previously, but I will clarify it here. Below you can see two examples of the DNA ends produced by restriction enzyme digestion and how to modify them for your plasmid design in SnapGene. If you are using a different software you can simply delete or add the bases manually if this function is not available. Once your fragment is modified it can be copied into the plasmid sequence in the correct position.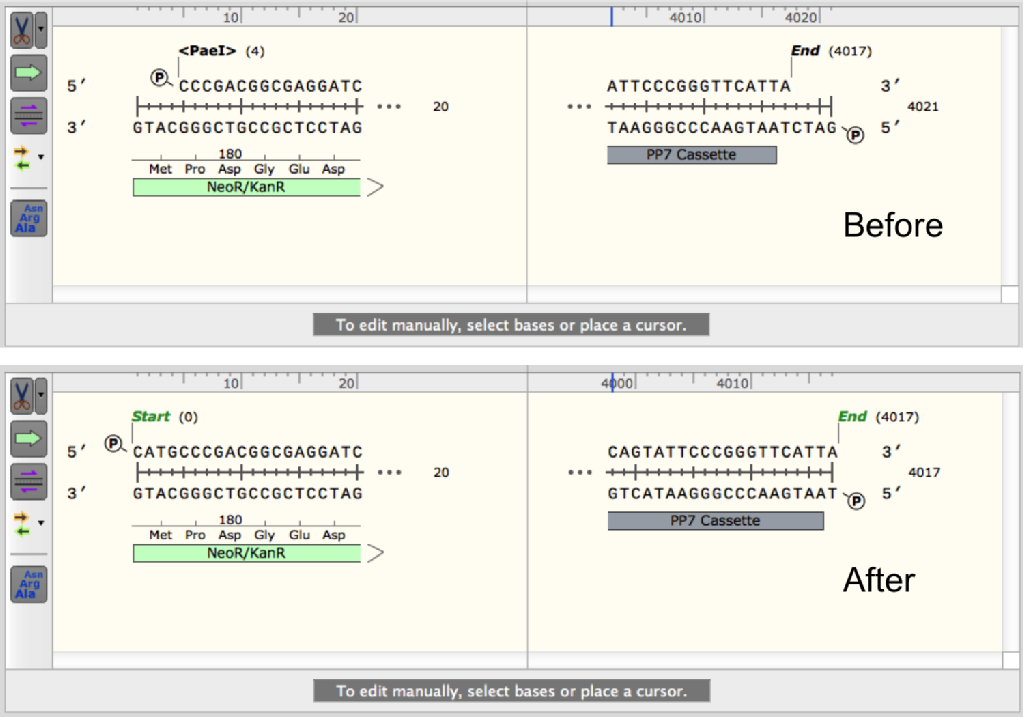 From your plasmid map you can now design your PCR primers for the fragments adjacent to restriction fragments. Using the same process as previously, first define a binding region for your primer in the PCR fragment, try to keep this as small as possible without sacrificing too much Tm as we want to keep the cost of the primer down. For AT rich fragments such as promoter regions this may be difficult and ordering a longer primer may be necessary. Once you have your binding region, extend the 5' end of your primer into the restriction fragment by 40 bp as shown below.
From your plasmid map you can now design your PCR primers for the fragments adjacent to restriction fragments. Using the same process as previously, first define a binding region for your primer in the PCR fragment, try to keep this as small as possible without sacrificing too much Tm as we want to keep the cost of the primer down. For AT rich fragments such as promoter regions this may be difficult and ordering a longer primer may be necessary. Once you have your binding region, extend the 5' end of your primer into the restriction fragment by 40 bp as shown below.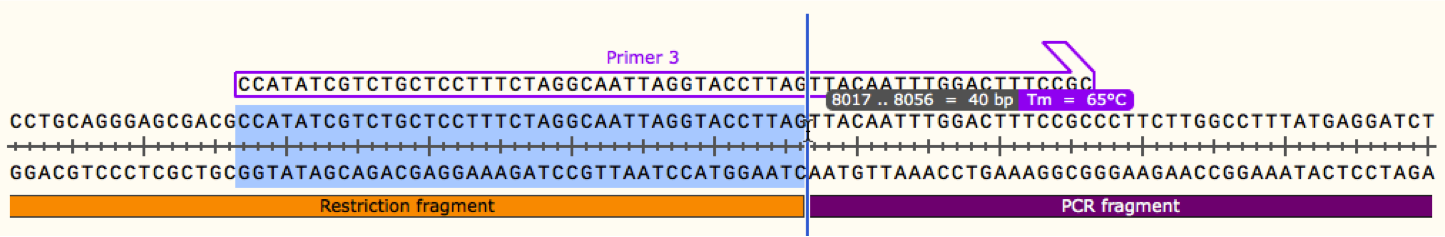 We now have a sufficient overlap to continue with the gibson reaction while incorporating the restriction fragment. Remember to repeat this process with all PCR-restriction joints to give sufficient overlaps throughout the plasmid.
We now have a sufficient overlap to continue with the gibson reaction while incorporating the restriction fragment. Remember to repeat this process with all PCR-restriction joints to give sufficient overlaps throughout the plasmid.
Final Step - Assembly
One you have generated your DNA fragments using your designed PCR primers and chosen restriction enzymes, you are ready to assemble your plasmid. You should first purify your fragments from the PCR/digestion reaction, either using a gel extraction kit or PCR purification columns. Remember when using PCR purification that you will also purify any template plasmid you used, so you should Dpn1 digest your product first to remove the methylated DNA. After purification, you must combine your fragments in the gibson assembly enzyme mix. There is a commercial kit available from New England Biolabs that provides pre-mixed gibson assembly enzymes and buffers that is also available with competent cells for transformation. However if you want a cheaper option, the mix can be made in the lab by yourself. The details for the homemade master mix can be found here along with the protocol for assembly of fragments. We use the second listed method, using the 1.33x master mix in 15ul aliquots, adding 5ul of DNA and incubating for 1 hour at 50oC followed by standard bacterial transformation into chemically competent cells.
Summary
Gibson assembly far out-performs standard restriction cloning when it comes to joining more than one fragment + backbone. In our lab we have successfully joined 5 fragments, 4 PCR or restriction fragments + a restriction digested backbone, with fragment sizes up to 5kb, although larger fragments should be possible. The design principles outlined above show how each fragment type should be treated and incorporated into your plasmid design with minimal cost. This guide deals only with fragment incorporation into plasmids, but the gibson procedure can also be used in other ways. The gibson assembly process can essentially be used for any type of homologous end joining. For example using a single primer set and plasmid, you can introduce mutations at any point in your plasmid, by changing the sequence in your primer overlaps and adding the PCR product (after Dpn1 digest) to the gibson mix, where it will be rejoined with the modifications. Gibson assembly can therefore replace most, if not all, of the current molecular cloning techniques being used in the lab today.