
Calculations
GIPAW |
Density Matrix Calculations |
QuadFit |
GIPAW DFT calculations of NMR parameters
The Gauge-Including Projector Augmented Wave (GIPAW) method that was presented by Pickard and Mauri in 2001 (Pickard 2001) enables the calculation of NMR parameters for periodic solids. The Warwick group is part of the EPSRC-funded collaborative computational project for NMR crystallography (CCPNC). Together with the CASTEP developers, Chris Pickard (UCL) and Jonathan Yates (Oxford), CCPNC and more recently also on our own, we have applied the GIPAW method to:
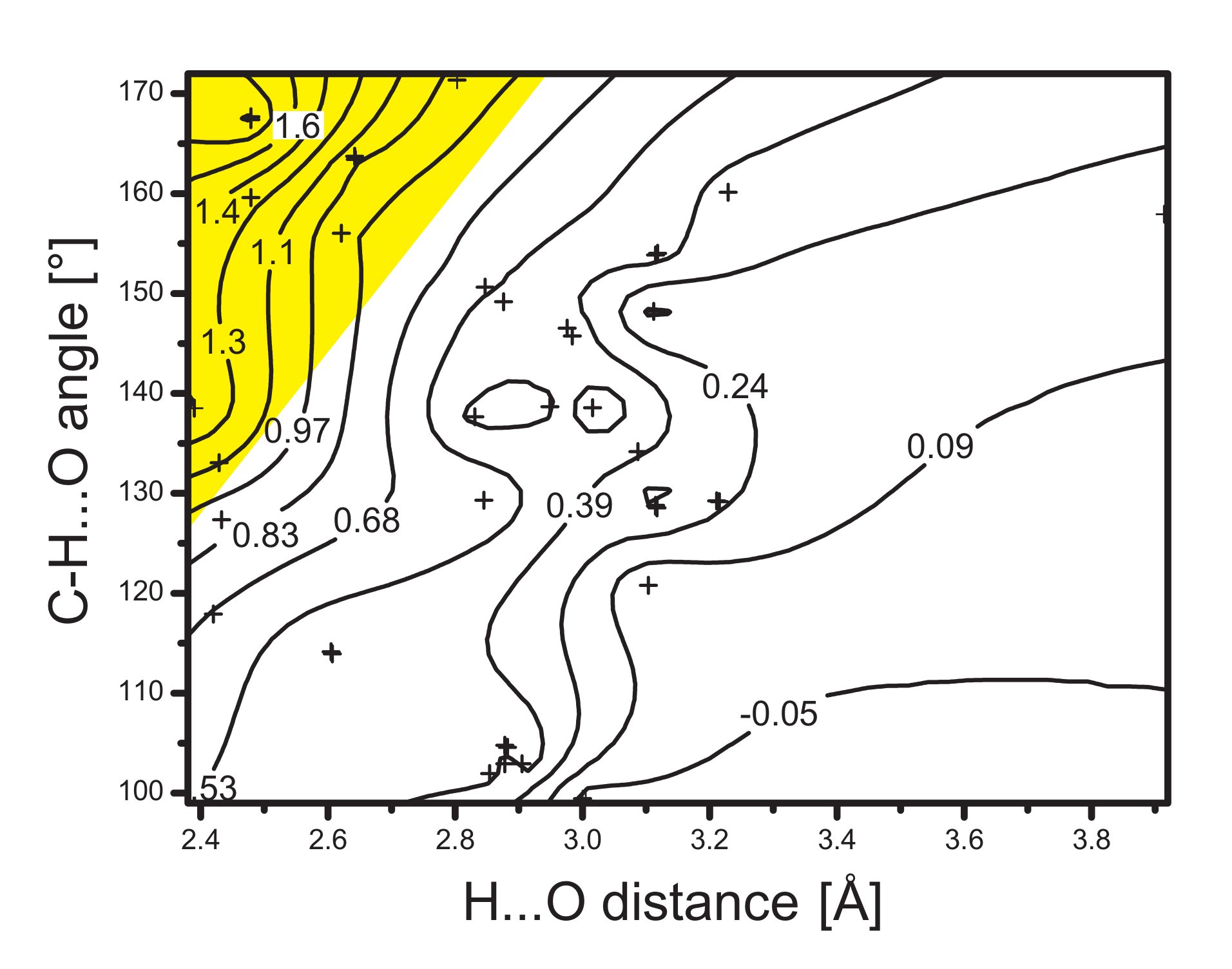
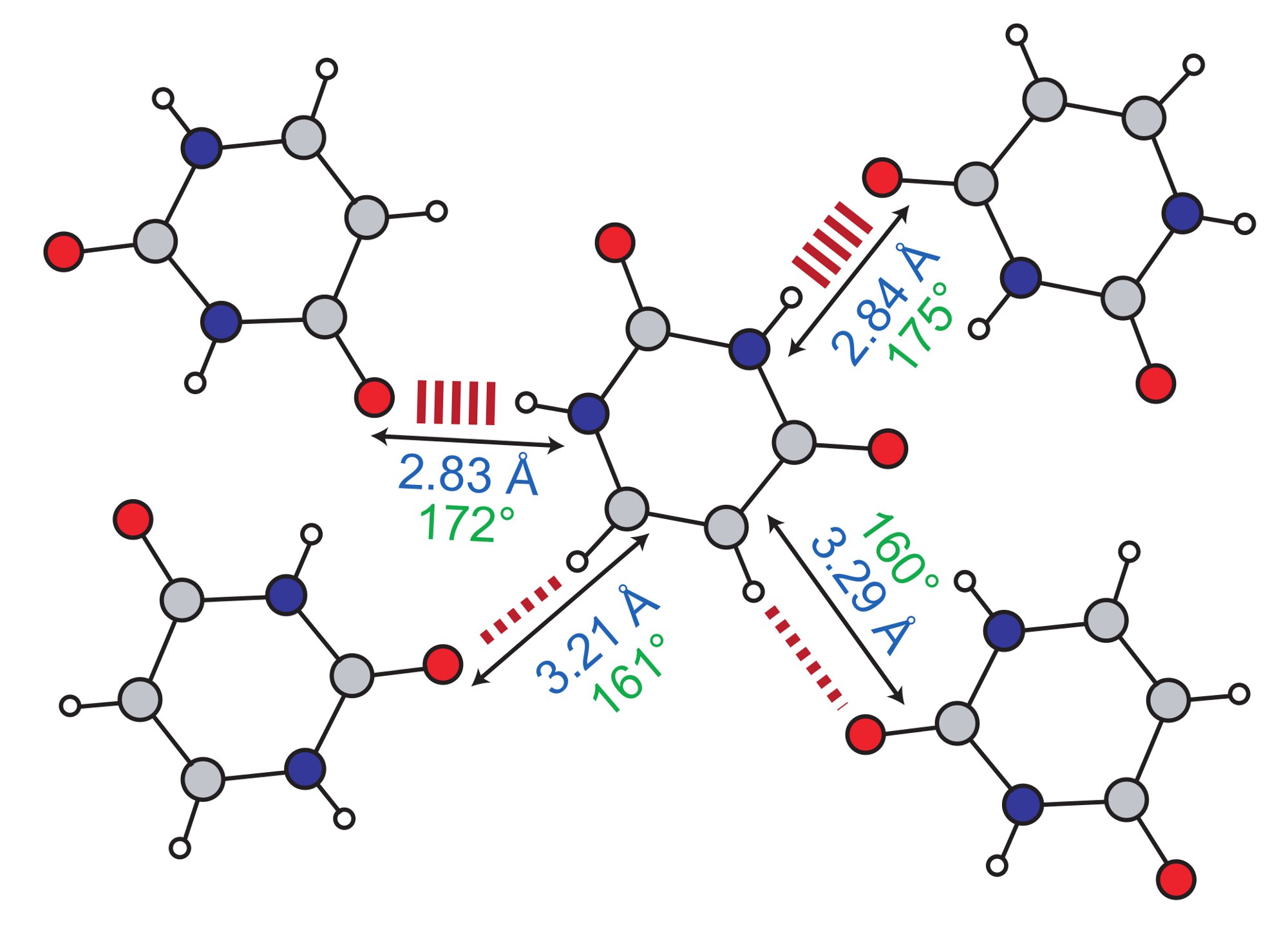
(i) Weak Hydrogen Bonding
|
|
|
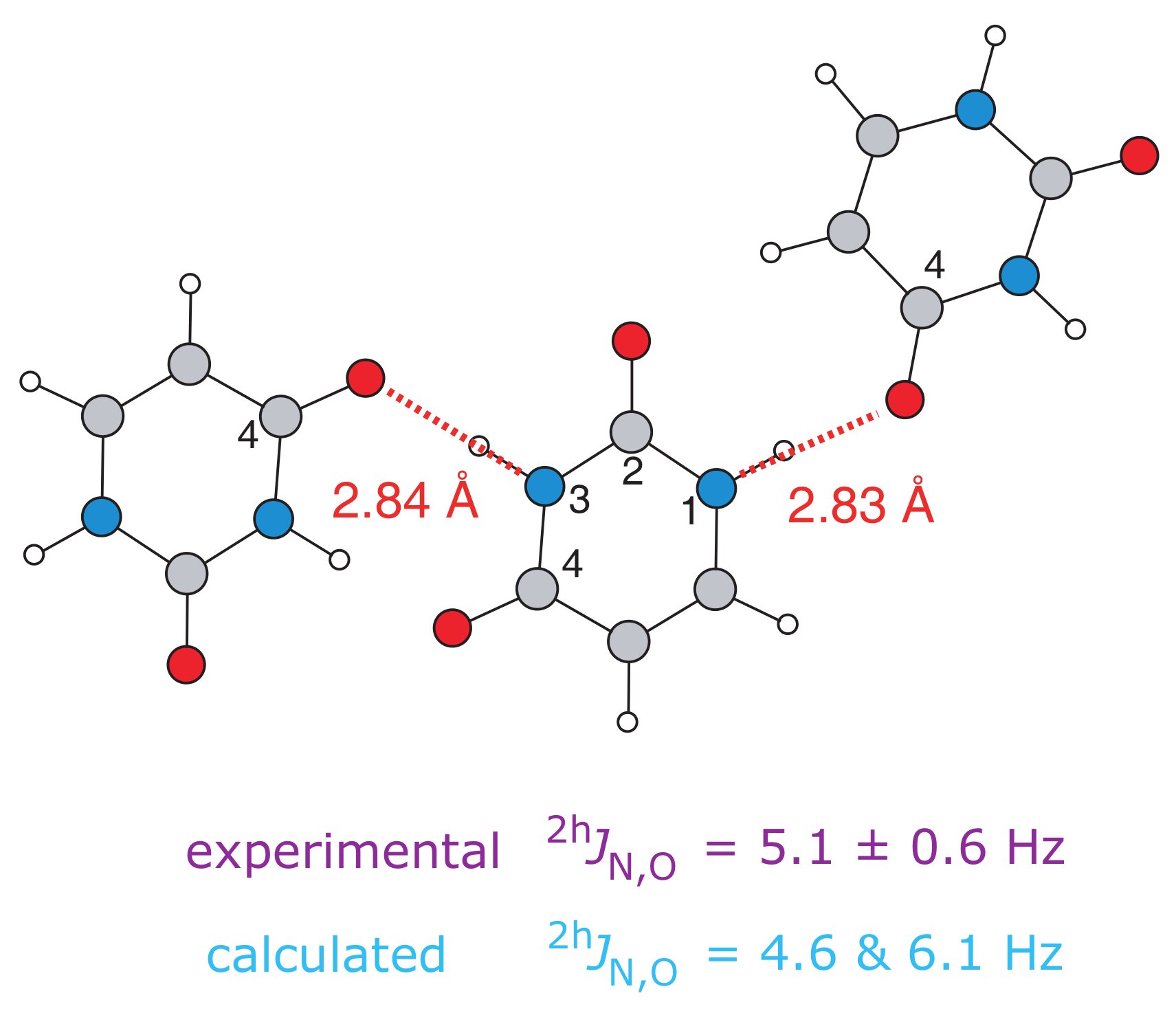
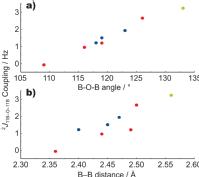
(ii) J couplingsExcellent agreement between calculation and experiment has been observed for 15N-15N (Joyce 2008) and 15N-17O (Hung 2009) hydrogen-bond mediated 2hJ couplings exhibited by organic molecules. Recently, calculations of 2J 11B-11B couplings in lithium borates have been presented, whereby their small values are correlated with small B-O-B angles (Barrow 2011). |
|
(iii) CH-π InteractionsThe method of comparing chemical shifts calculated for the full crystal structure and for isolated molecules can be used to probe CH-π interactions, notably the effect of ring currents associated with aromatic moieties, as illustrated for the crystal packing adopted in the γ polymorph of the pharmaceutical molecule, indomethacin (Bradley 2011). |
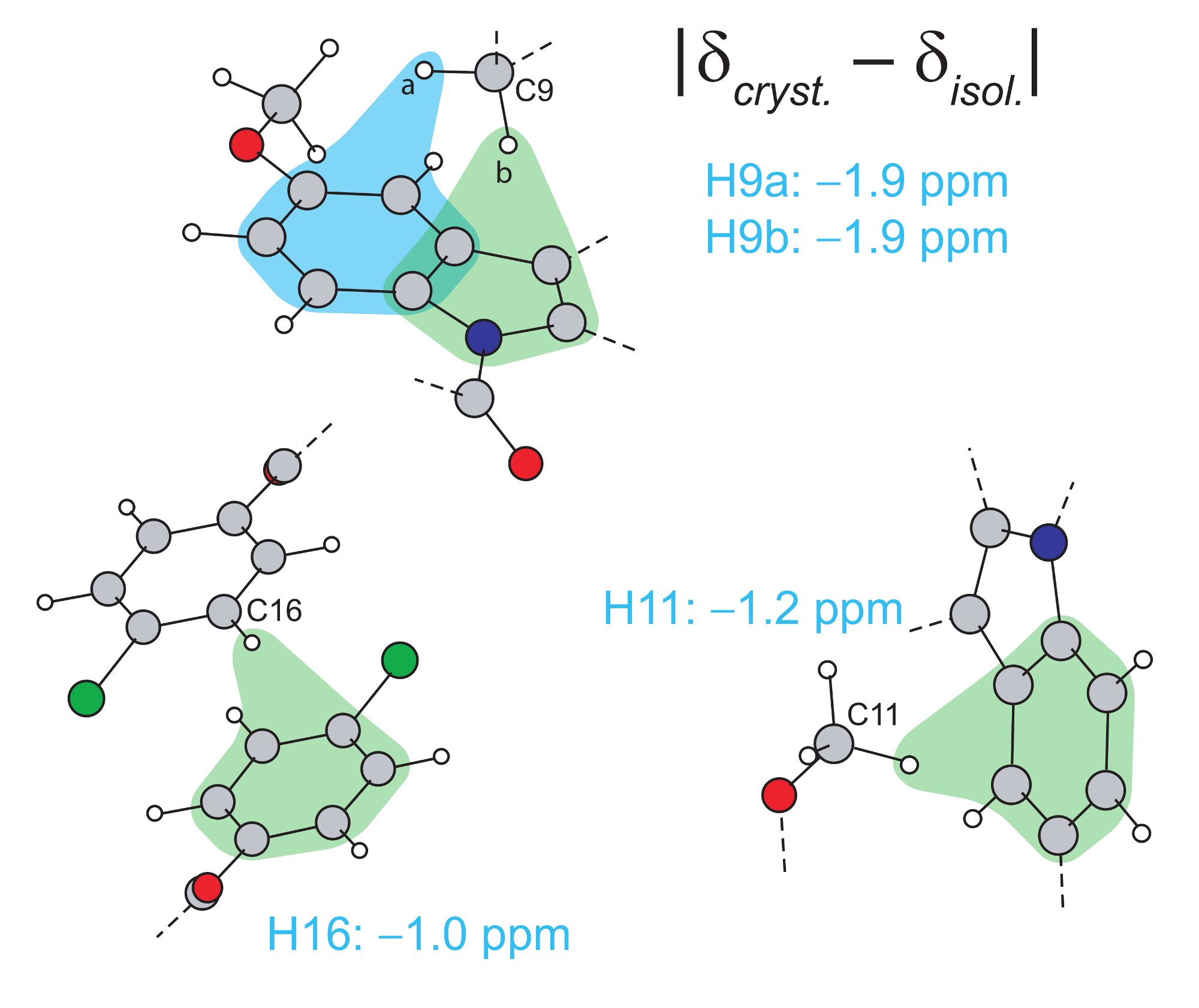 |
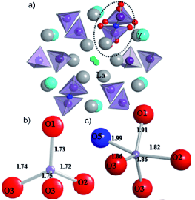 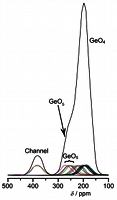 |
(iv) Identifying O speciation in rare earth and transition metals oxide materialsGIPAW DFT computational methods have been crucial in the identification of interstitial O species responsible for O conduction in rare earth apatite solid oxide fuel cell (SOFC) membrane materials (Panchmatia 2011). These methods are now being extended to O deficient transition metal perovskite materials used as SOFC electrodes, where disordered A site, B site and O vacancies are fundamental to their function. |
|
|
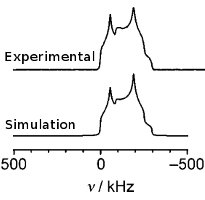 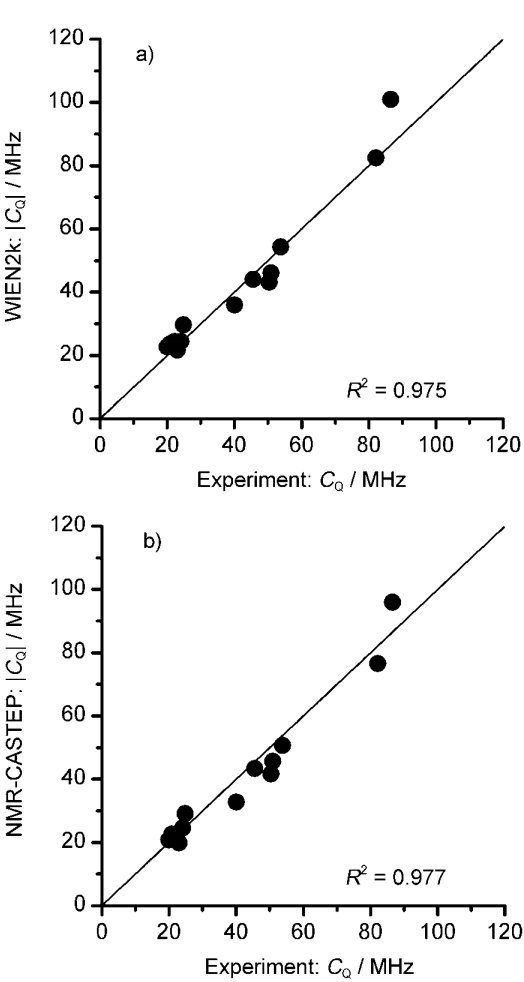 |
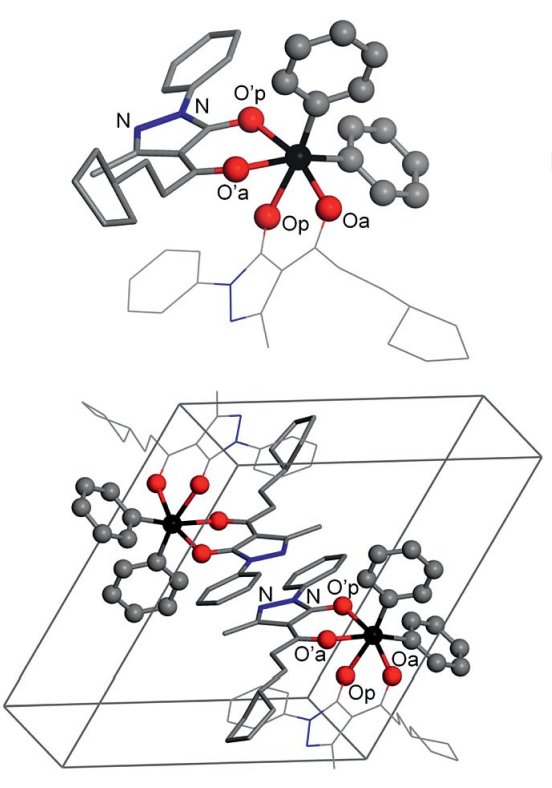 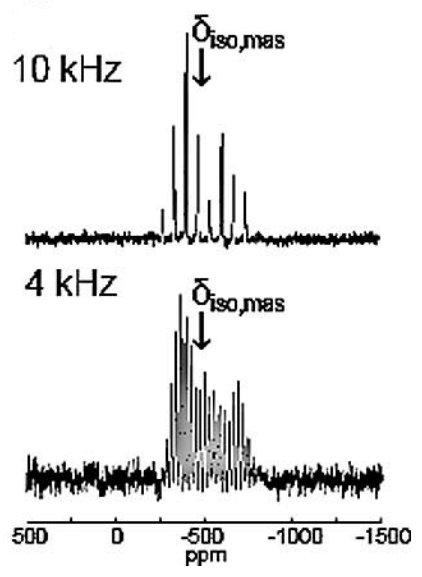 |
(vi) Metal complexes for biological and clinical applicationsStudies into diorganotin(IV) β-diketonates which demonstrate clear insecticidal, antifungal and antitumour properties have shown that this family of compounds adopts a regular trans octahedral geometry. More recent work (Caruso 2012) utilising 119Sn MAS NMR and GIPAW DFT computation has reported unprecedented cis arrangements relating to disordered phenyl substituents within this series. This occurrence highlights some important implications regarding the reactivity and flexibility of these complexes when considering their implementation in biological and clinical applications. |
Density-Matrix Calculations
|
The development and application of advanced solid-state NMR pulse sequences is supported by performing multi-spin density-matrix simulations. In particular, the Warwick group, in collaboration with Claudiu Filip's group in Cluj have used SPINEVOLUTION to investigate the structural information that can be extracted from CHHC (Aluas 2009) and 1H DQ (Bradley 2009) build-up curves. |
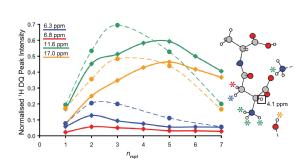 |
QuadFit
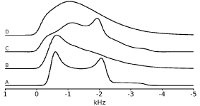 |
QuadFit is a platform independent program for simulating distributions of Quadrupolar interactions. QuadFit's calculations are performed in the frequency domain to maximise performance. QuadFit was written and is maintained by Dr Thomas Kemp. Contained within the QuadFit pages is various documentation including instructions on using the program. QuadFit was designed to allow simulation of NMR spectra from materials which have atomic disorder resulting in a distribution of NMR interaction parameters. The information which can be extracted from the spectra gives a measure of the disorder of the material and combined with multiple field data can allow the deconvolution of multiple overlapping sites. |