
QuadFit Interface Description
Starting QuadFit
Once java is installed QuadFit can be started in several different ways.
Windows:- Double click on the icon and if Java and Windows is set up correctly the starting window will apear. If it does not then open a command prompt (open "Run" and type "cmd"), use cd (changed directory) to navigate to the location of quadfit and type "java -jar quadfit.jar" and the program should start.
Linux:- From a terminal window type "java -jar quadfit.jar".
Note:- If you are intending to do particularly complex distributions which will require a lot of memory the standard limit memory usage limit in java may not be large enough, to get around this problem the -Xmx modifier can be used like "java -jar -Xmx512m QuadFit.jar" this will alow the program to use 512Mb of RAM.
Initial Window

In this initial window the Larmour Frequency in MHz, nuclear spin and the resolution which the program runs at can be set. The initial value of the resolution is the default and the program works best at this however it can be changed if required. Once the desired values have been entered, click the "OK" button to activate the main program.
Main Window

Interface Description
The interface is split into various different panels.
A
Shows the individual simulates lines, total simulated line, spectra and difference spectra.
B
Three parameters which control the range over which the spectra and fit are displayed. The setting are L H Freq, R H Freq and Y Axis Scale. The two frequency settings can be changed to display the frequency range as required. The Y axis of the graph automatically scales to the largest feature detected. This can be scaled using the "Y Axis Scale", a setting of 2 will double the hight of the spectra displayed.
C
This shows which peak is currently selected and allows you to add and remove more peaks. The lowest number peak that can be selected is number 3 as number 0 is the spectra, 1 is the total fit and 2 is the difference spectra.
To add a new peak click on the "Add Peak" button and another peak will be added using the same parameters as the currently selected peak. The new peaks number will be shown in the "Peak Select" box and will be the highest numbered peak. The maximum number of peaks that can be used is 100.
Currently the "Delete Peak" button does not work at the moment, to effectivly delete a peak set it's intensity to 0.
To change which peak is selected either use the up and down keys to cycle through the peaks or type the desired peak number in the "Peak Select" box and press Return.
D
Shift Spectra Vertically allows for corrections for DC offset of the spectra, the units are in intensity for the spectra so very large numbers can be required. To use this type in the desired number and press return.
E
"Load Spectra" This button allows you to load spectra into the program for fitting. QuadFit will load in Bruker data where the data has been saved to a text file without the imaginary data and also in a two columns format. The two column format must have two columns of number, seperated by two spaces, the first column should be shifts in ppm going from high to low and the second column intensities at that shift. Only two numbers per line should be permitted and there should be an even number of points. Please note that a lot of windows based programs like Excel do not show you exactly what is in a file. My program of choice for rearranging the contents of files is Gnumeric.![]()
"Don't Show Noise" When clicked this button changes between "Don't Show Noise" and "Show Noise", like all of the buttons which change in the program they show what is being done at that time. When changed to "Show Noise" the difference spectra (total fit subtract the spectra) between the total fit and the spectra will be displayed.
"Save" will open a dialogue box asking where to save all the individual lines, total fit, difference spectra and the spectra in a text file. The data saved at the time will only be that displayed on the screen at the time. The text file is in a format of columns of space separated data. The order of the columns is:- spectra shifts in ppm, space, spectra intensities, space, total fit shifts in ppm, space, total fit intensities, space, difference spectra shifts in ppm, space, difference spectra intensities, space, line 3 shifts in ppm, space, line 3 intensities, space, line 4 shifts in ppm,space, line 4 intensities, etc.
"Iterate" will open a itteration dialogue box which will be explained later.
F
"Intensity of Line" changes the intensity of the currently selected line. The shortcut key is "h".
"Shift of Line (ppm)" changes the isotropic shift of the currently selected line. The shortcut key is "s".
G
"norm. resid. per pt." shows the normalised residual per point. This is calculated by first calculating the root mean squared of the difference spectra and is then normalised by how many points there are in the experimental spectra and the maximum intensity of the experimental spectra. This results in a number which has the same value for the same quality of fit irrespective of either horizontal or verticle scale.
"Width of CS Dist." and "No lines to calc" are not used and will be removed at some point.
Progress Bar. At the bottom of this panel is a progress bar which shows how far through a calculation QuadFit is.
H
This panel contains the information on how the program will calculate the simulations. The initial values are chosen with the idea of providing a fair comprimise between calculation quality and speed.
"No calc steps" changes the number of steps each face is split into during the alderman grant interpolation. The higher the number the better quality the simulation, however, the longer the simulation takes. This number must be an odd number!
"No freq steps =2^n" changes the number of frequency steps used during the simulation process. Again the higher the number the better the simulation but the longer it takes. This number must be a power of 2!
"Lowest freq to calc" and "Highest freq to calc" alow the range (in kHz) over which the simulation is calculated over to be set. This number should be set atleast twice as wide as the line shape being calculated, however if it is set too wide the number of frequency steps will have to be increased to keep a good quality like.
I
This panel contains the information which the program uses to calculate the lines which a distribution will be calculated from. As the type of line changes the parameter names change but the are relatively self explanitary.
"Min. Quad. Coup." minimum quadrupole coupling constant in MHz. If "No. Quad. Dist. Calcs."=0 the short cut key is "u".
"Min. Asym." minimum asymmetry parameter between 0 and 1. If "No Asym. Dist. Calcs."=0 the short cut key is "o"
"Max. Quad. Coup." maximum quadrupole coupling constant in MHz.
"Max. Asym." maximum asymmetry parameter between 0 and 1.
"No. Quad. Dist. Calcs." number of steps to calculate for the quadrupole distribution.
"No Asym. Dist. Calcs." number of steps to claculate for the asymmetry distribution.
If no distribution is required then enter the values wanted for the calculation in the minimum boxes and make sure that the number of steps is to 0.
QuadFit works by calculating each line required for a distribution between the min and max values with the number of steps stated and then saving each line shape internally. QuadFit will then add the intensities of the lines depending on the distribution chosen. This results in a much more fluid way of working as once the lines have been calculated changes in the line shape can be calculated very quickly if the distribution is changed. Care needs to be taken when calculating distributions in both asymmetry and Cq as QuadFit will calculate very point of the two dimensional distribution, and with 100 steps in each dimension this is 10,000 points and will take approximately 20 minutes depending on the quality of calculation.
J
"Broadening (Hz)" Allows broadening of the currently selected line. The shortcut key is "b"
"0=Gaus., 1=Lorent." Allows the broadening to the currently selected line to be changed continuously between Gaussian=0 and Lorentzian=1.
"MAS" allows changing of the line type being calculated. The initial line is a second order line under infinite speed MAS. The cycle is "MAS", "Static" which is a second order quadrupolar line whilst static, "CSA (static) which is a CSA line whilst static and the names in panel "I" change, "CSA+Quad (static)" which is combined CSA and Quadrupolar interactions whilst static and an extra dialogue box opens which will be discussed later, and finally kroniker which gives a sharp one point wide line which can then be broadened if a gaussian/lorentzian line is required.
K
"Integrated Intensity" shows the integrated intensity of the currently selected line as a percentage of the whole fit.
L
"Calculate" button allows the program to recalculate after the parameters have been changed, this can be activated by pressing "Return". The exact recalculation that is done is dependant on which values have changed, simple changes like the change in intensity, isotropic shift, center and width of a distribution and type of distribution can be done almost instantly. Changes like changing the range over which a distribution is calculated can take much longer.
M
This panel controls the type and shape of the distributions used on the interaction parameters. When the number of steps in a dimension is set to zero the distribution in that dimension is ignored. Usually the range of lines to be distributed over are calculated first and after which the display shows no line as the initial values in this panel ask for a distribution centered around zero with no width. To solve this change the center and width to appropriate values. Although the program allows you to set the width greater than the center care needs to be taken that the number used make sense as negative quadrupolar interaction parameters are meaningless. Care also needs to taken so that the distribution does not extend over the range it has been asked to calculate over.
"Dist. Type" this button cycles throught the different types of distribution that can be used "Gaussian", "Czjzek" and "Flat". Under a gaussian distribution the center and width of the distribution in both Cq and asymmetry can be set. Under Czjzek distribution two values can be set which are d and sigma. Sigma sets the width and centre of the distribution while lower values of d imply local geometrical constraint.
Iteration Window
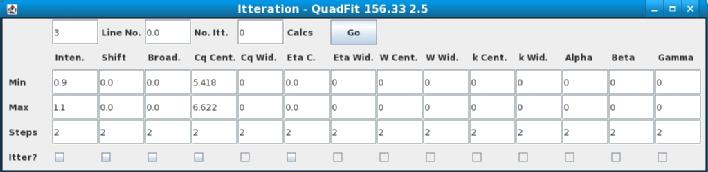
This window is for itterating the parameters of the line to gain the best fit. For each parameter the minimum, maximum and number of steps can be set. There is also a tick box to activate the itteration of that variable. When opened the minimum and maximum values of the variable which can be itterated are set to 10% above and below the currently used value.
The itteration section of the program is currently still very much under development and if you come across any problems please contact me.
"Line No." shows which line is currently selected, either changing the number in the box and pressing return or using the up and down buttons allows you to switch between the lines. The program will remember the settings as you change through lines.
"No. Itt." this can be set to allow the itteration algorithm to run multiple times. Once the itteration has run for the first time it will choose the parameters which give the best result and then use the points on either side for the minimum and maximum of the next itteration. The most efficient way of running an itteration is to use multiple cycles of the algorithm with relatively small amount of steps.
"Calcs" shows how many lines need to be calculted. Be careful that this number does not get too large as the itteration could use a lot of memory and take a lot of time.
"Go" button sets the itteration going.
Combined Quadrupolar and CSA Parameters Window

This window allows the setting of interaction parameters for simulation of combined and quadrupolar and CSA interactions. The window also allows for setting of distributions of the interaction. The line shape simulated depends on not just the parameters which define each interaction but also the relative orientation of the two tensors. The orientations of the two tensors are defined by euler angles and are defined below..
The calculation of the combined quadrupolar and CSA interactions will include all of the outer transitions if the window width is large enough that they can be seen to have an effect. An upshot of this is that only the quadrupolar interaction can be calculated with all of the transitions by leaving the CSA interaction parameters at zero.
"Alpha (deg)" Short cut key "j". Alpha is a rotation of the EFG tensor around the Z axis.
"Beta (deg)" Short cut key "k". Beta is a rotation of the EFG tensor which has already been rotated by Alpha around the Y' axis.
"Gamma" (deg)" Short cutkey "L". Gamma is a rotation of the EFG tensor which has already been rotated by Alpha and Beta aroung the Z'' axis.
"Span (ppm)" is the span of the CSA interaction. Short cut key for the minimum (if no.=0) or center (if no.>0) is "E" and for the width is "R". The span is defined as W=d11-d33 W>=0.
"Skew" is the skew of the CSA interaction. Short cut key for the minimum (if no.=0) or center (if no.>0) is "T" and for the width is "Y". The skew is defined as k=3(d22-diso)/W -1<=k<=+1.
"Coup. (MHz) is the Cq of the quadrupolar interaction. Short cut key for minimum (if no.=0) or center (if no.>0) is "U" and for the width is "I".
"Asym Quad" is the asymmetry parameter for the quadrupolar interactions. Short cut key for the minimum (if no.=0) and center (if no.>0) is "O" and for the width is "P".
"min" allows you to set the minimum value calculated for a distribution. If the number of steps ("no.") is set to zero a single line in that dimension of the distribution is produced with the value in this box.
"max" allows you to set the maximum value calculated for a distribution.
"no." allows the setting of the number of steps between the minimum and maximum values set. If this value is 0 then a single line in that dimension of the distribution is produced using the minimum value set. Care needs to be taken with the number of steps as it can easily reach 108 calculations if 100 steps in each dimension is taken which would take >116day.
"cent." allows the setting of the center of the distribution. If this value is not set before the line shapes are calculated no line shape will be displayed as a distribution of center 0 and width 0 has been requested.
"width" allows the setting of the width of the distribution. Although this value can be set larger than the center it is meaningless to do so as it will give a skewed distribution. Care needs to be taken that the choice of min, max, center and width does not result in a truncated distribution.