
Peptidoglycan
Enzymes Involved in Bacterial Peptidoglycan Biosynthesis
The assembly of the peptidoglycan layer of bacterial cell walls is the target for a number of clinically used antibiotics such as the penicillins, the cephalosporins, and vancomycin. The rapid acceleration of bacterial resistance to these antibiotics over the last 10 years has severe implications for the medical community, and underlines the need to find new types of antibiotics [1]. The assembly of the peptidoglycan layer of bacterial cell walls represents a good target for antibiotic action.
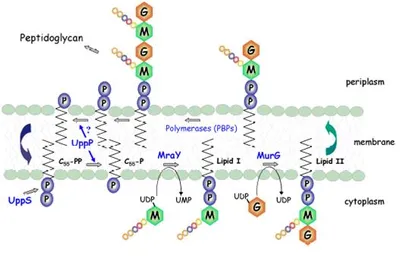
Peptidoglycan biosynthesis occurs via the assembly of a cytoplasmic precursor UDPMurNAc-L-Ala--D-Glu-m-DAP-D-Ala-D-Ala by a series of ATP-dependent ligases, followed by the translocation of the MurNAc-pentapeptide across the cytoplasmic membrane by means of a lipid carrier, undecaprenyl phosphate:
 |
|
Figure 1: The Membrane steps involved in peptidoglycan biosynthesis. MurNAc (M), |
1. Phospho-MurNAc-pentapeptide translocase
Phospho-MurNAc-pentapeptide translocase catalyses the attachment of peptidoglycan precursor UDPMurNAc-L-Ala-D-Glu-m-DAP-D-Ala-D-Ala to a membrane carrier undecaprenyl phosphate. This is the first step of a cycle of reactions involved in transporting the peptidoglycan precursors across the cytoplasmic membrane and assembling into cell wall. This enzyme is an integral membrane protein, which presents particular problems as regards enzyme purification. The enzyme has been solubilised by detergents, and purification of the enzyme has commenced. Three enzyme assays have been developed, including a novel continuous fluorescent assay. Using this assay, a peptide nucleoside natural product mureidomycin A has been identified as a slow-binding inhibitor of this enzyme, with Ki= 38 nM and Ki* = 2 nM [2]. In contrast, the well-known tunicamycin is a reversible, competitive inhibitor with Ki= 0.55 µM [3].
The molecular mechanism of action of mureidomycin A has been studied in our research group. MrdA contains an unusual enamide functional group, which could be involved in enzyme inhibition.
Enamide-containing model compounds have been synthesised. A tetrahydrofuran-based enamide was found to be reactive and unstable, however a uridine-based enamide was found to be stable and unreactive, similar to the natural product, and did not inhibit the enzyme. Thus, although the enamide can in principle undergo the above mechanism, there is no evidence that it does in this case [4].
Structure-function studies on mureidomyain A itself have revealed that several functional groups in the molecule appear to contrbute to the binding affinity. Synthetic derivatives in which the amino terminus and the carboxyl terminus are attached to a uridine nucleoside have shown activity as inhibitors, implying that the N-terminal and C-terminal ends of the peptide chain are important for binding [5].
A series of uridinyl dipeptides mimicking the amino-terminal arm of mureidomycin A have been synthesised, and tested for translocase I inhibition and antibacterial activity. Uridine 5’- L-alanyl-N-methyl--alanyl ester showed the most potent enzyme inhibition, and was active against Pseudomonas putida at 100 µg/ml. Compounds containing N-methyl-beta-alanine showed cis and trans amide rotamers, furthermore the N-methyl group was required for high biological activity, suggesting that the role of the N-methyl group may be to form an active conformation via a cis-amide geometry. Enzyme inhibition was competitive with Mg2+. Our hypothesis is therefore that the amino terminus of MrdA binds in place of Mg2+ at the translocase I active site, and is positioned by a cis-amide bond [6].
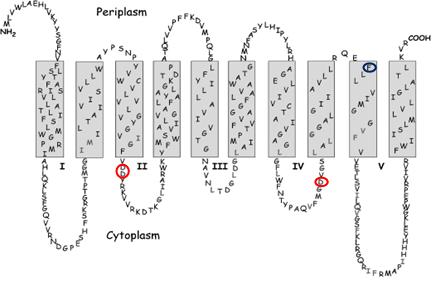 |
| Figure 2: Predicted membrane structure of E. coli MraY. Red circles mark the location of the active site residues. The blue circle marks the site of the mutation that confers resistance to protein E. |
E. coli translocase I has been expressed as a C-terminal His6 fusion protein, and has been detected by Western blot analysis. Secondary structural predictions indicate that this membrane protein has ten transmembrane helices. There is literature evidence for a two-step catalytic mechanism, proceeding via a covalent intermediate. Sequence alignments indicate only three completely conserved residues that might function as a catalytic nucleophile: Asp-115, Asp-116, and Asp-267, each positioned on intracellular loops. It is proposed that Asp-115 and Asp-116 form a Mg2+ binding site, and that Asp-267 may function as the nucleophile. Replacement of each residue by site-directed mutagenesis gives inactive enzyme, indicating that these residues are essential for catalytic activity [7].
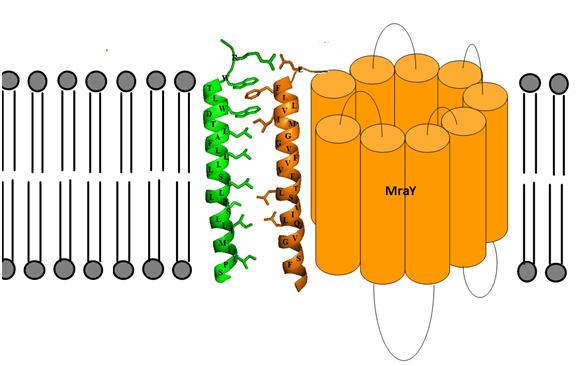
MraY has also been shown to be the target for an antibacterial E protein from bacteriophage phiX174. A synthetic 37 amino acid peptide containing the transmembrane domain of E has been shown to inhibit membrane-bound MraY, but not solubilised MraY, suggesting that E forms a protein-protein interaction with MraY in the membrane, that disrupts other productive protein-protein interactions in vivo involving MraY. We have developed a model for the possible transmembrane interactions between protein E’s transmembrane helix and MraY’s helix 9. Our model elucidates the possible formation of aromatic binding and salt bridge electrostatic interaction between the two. Phe-288 can form aromatic π-bonding with Trp-4 and Trp-7 of protein E, while Glu-286 can form a salt bridge with Arg-3 of protein E [8].
 |
|
Figure 3: Predicted transmembrane interactions between Epep and MraY translocase. |
2. Glycosyltransferase MurG
MurG catalyses the transfer of a GlcNAc sugar from UDPGlcNAc to lipid intermediate I , forming lipid II, and represents an attractive antibacterial target. Using a fluorescently labelled substrate analogue, a FRET assay has been developed for MurG [9]. We are currently developing new synthetic inhibitors for MurG.
3. tRNA-dependent ligases MurM and MurN from Streptococcus pneumoniae
High-level penicillin resistance in Streptococcus pneumoniae is a serious clinical problem in a number of countries around Europe and the world. Highly resistant strains contain a dipeptide cross-link (Ala-Ala or Ser-Ala) in their peptidoglycan structure. The murMN genes responsible for insertion of the dipeptide cross-link have been identified, and the corresponding MurMN proteins are believed to add Ala or Ser from the respective aminoacyl tRNA onto lipid intermediate II of peptidoglycan assembly.
Since 2003 we have collaborated with Prof. Chris Dowson and Dr. David Roper (Department of Biological Sciences) to reconstitute the MurM and MurN reactions. We have recently reconstituted the MurM-catalysed reaction in vitro, and have kinetically characterised enzymes from penicillin-resistant and –susceptible strains of S. pneumoniae [10]. We have also synthesised the first inhibitor for the MurMN enzyme family, an adenosine phosphonate transition state analogue [11].
4. Development of new methods to study in vitro peptidoglycan biosynthesis
The late lipid-linked steps of peptidoglycan biosynthesis are very challenging to study, involving complex lipid-anchored disaccharide-peptide substrate, and membrane-bound enzymes. In collaboration with Prof. Steve Evans (Department of Physics, University of Leeds), we are developing new methods to deposit lipid intermediates I and II into tethered lipid bilayers. The lipid-linked steps could then be studied using surface plasmon resonance spectroscopy [12], fluorescence microscopy, and other surface science methods.
We have also developed methods for the incorporation of fluorescent labels into the peptide chain of the peptidoglycan precursor: either via chemical modification of the amino group sidechain of Lys/meso-DAP; or via incorporation of an unnatural D-Cys residue in place of D-Ala, and modification of the thiol sidechain of D-Cys [13]. We are currently developing these methods to study the late steps of peptidoglycan assembly.
5. Other enzymes of bacterial cell wall metabolism
D-Ala-D-Ala adding enzyme catalyses the ATP-dependent condensation of UDPMurNAc-L-Ala-D-Glu-m-DAP with the D-Ala-D-Ala dipeptide. We have purified the enzyme from Escherichia coli, and have synthesised a series of aminoalkylphosphinate transition state analogues. These analogues proved to be modest competitive inhibitors for the enzyme, the best inhibitor showing Ki= 200 µM [14].
The peptidoglycan hydrolases are a family of enzymes involved in the breakdown and re-assembly of peptidoglycan required for bacterial growth. The consequences of inhibition of such enzymes are not yet clear, in view of the lack of molecular studies on these enzymes. We are developing new synthetic substrates for MurNAc-L-Ala amidase, which catalyses the hydrolysis of the MurNAc-L-Ala linkage, in order to allow the design and synthesis of rational inhibitors for this enzyme [15,16].
References
1. T.D.H. Bugg & C.T. Walsh, Nat. Prod. Rep., 9, 199-215 (1992)
2. P.E. Brandish, M. Burnham, J.T. Lonsdale, R. Southgate, M. Inukai, & T.D.H. Bugg, J. Biol. Chem., 271, 7609-7614 (1996)
3. P.E. Brandish, K. Kimura, M. Inukai, R. Southgate, J.T. Lonsdale and T.D.H. Bugg, Antimicrob. Agents Chemother., 40, 1640-1644 (1996)
4. C.A. Gentle and T.D.H. Bugg, J. Chem. Soc. Perkin Trans 1, 1279-1286 (1999)
5. C.A. Gentle, S.A. Harrison, M. Inukai and T.D.H. Bugg, J. Chem. Soc. Perkin Trans 1, 1287-1294 (1999)
6. N.I. Howard and T.D.H. Bugg, Bio-Organic and Medicinal Chemistry, 11, 3083-3099 (2003)
7. A.J. Lloyd, P.E. Brandish, A.M. Gilbey, and T.D.H. Bugg, J. Bacteriol., 186, 1747-1757 (2004)
8. S. Mendel, J.M. Holbourn, J.A. Schouten and T.D.H. Bugg. Microbiology, 152, 2959-2967 (2006)
9. J.J. Li and T.D.H. Bugg,J. Chem. Soc. Chem. Commun., 182-183 (2004)
10. A.J. Lloyd, A.M. Gilbey, A.M. Blewett, G. De Pascale, A. El Zoeiby, R.C. Levesque, A.C. Catherwood, A. Tomasz, T.D.H. Bugg, D.I. Roper, C.G. Dowson, J. Biol.Chem., 283, 6402-6417 (2008)
11. E. Cressina, A.J. Lloyd, G. De Pascale, D.I. Roper, C.G. Dowson, and T.D.H. Bugg, Bio-Org. Med. Chem. Lett., 17, 4654-4656 (2007)
12. M. J. Spencelayh, Y. Cheng, R. J. Bushby, T.D.H. Bugg, J-J. Li, P. J. F. Henderson, J. O'Reilly, S. D. Evans,Angewandte Chemie Intl. Ed., 45, 2111-2116 (2006)
13. J.A. Schouten, S. Bagga, A.J. Lloyd, G. De Pascale, C.G. Dowson, D.I. Roper, and T.D.H. Bugg, Molecular Biosystems, 2, 484-491 (2006)
14. D.J. Miller, S.M. Hammond, D. Anderluzzi, and T.D.H. Bugg, J. Chem. Soc. Perkin Trans. 1, 131-142 (1998)
15. R.L. Harding and T.D.H. Bugg,Tetrahedron Letters, 41, 2729-2731 (2000)
16. R.L. Harding, J. Henshaw, J. Tilling, and T.D.H. Bugg, J. Chem. Soc. Perkin Trans. 1, 1714-1722 (2002)