
Additional LiBiNorm options
The following additional options may be available in development versions of LiBiNorm. A ✔ symbol indicates that they are enabled in the current version
Set initial values from a file
(Enabled by compling with INITIAL_VALUES defined)
Adds an option which allows the initial values for the MCMC stage to be preset from values in a file using the -i <filename> option. The format of the file is as follows:
A -0.4246024 0.6783496
B -0.549208 1.12126 -4.833895 -3.512791
C 0.303502 0.2770251 -4.951562
D -0.7436946 1.825406 -3.74649 -3.272332
E -0.4013753 1.874199 -1.150785 -4.402464
BD -0.132114 2.036754 -3.982542 -2.990412 0.8643603
Use genes/transcripts from a list
(Enabled by compling with USE_GENES_FROM_GENELIST defined)
Adds a -g <filename> (S F) option which specifies that only the genes or transcripts listed in the specified file should be used and all other genes and transcripts described in the gtf or gff file should be ignored. The optional S and F parameters allow just the genes from position S to F to be used.
Specify seed for random number generator (model mode only)
(Enabled by compiling with SELECT_READ_SEED defined)
In each run of LiBiNorm a set of up to 100 forward and reverse reads are selected for each gene. This is done with a random number generator that is initialised with a random seed. Adds an option that allows the seed to be explicitly specified using the -y N option in order to examine the robustness of the algorithm, by specifically selecting different sets of reads for the parameter determination.
Output debug messages
(Enabled by compiling with OUTPUT_DEBUG_MESSAGES defined)
Adds a -w option which causes the program to output additional debug messages.
Pause at end
(Enabled by compiling with PAUSE_AT_END_OPTION defined)
Adds a -x option which causes the program to pause at the end waiting for user input rather than simply finishing. This can be useful for debugging.
Output additional genome information
(Enabled by compiling with OUTPUT_FEATURE_DATA defined)
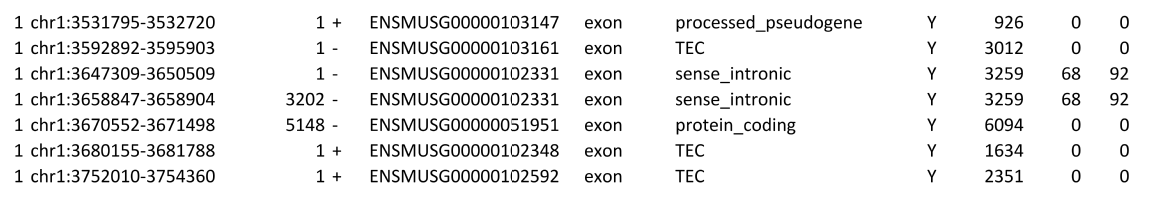
If the -c option is being used, then a further file, <countFilenamePrefix>_genome.txt, is created that provides more information about the information obtained from the feature file. This shows each of the regions that have been identified from the gff or gtf file.
The third and fourth column gives the start position of the exon within the RNA transcript and the strandedness of the gene. The final four columns indicate whether the region is part of a gene/transcript being used for parameter estimation, the length of the transcript and the number of forward and reverse reads associated with the whole transcript (not the individual exons).
Print MCMC run data - model mode only
(Enabled by compiling with PRINT_MCMC_RUN_DATA)
As well as the normal three results data files, this option prints out files for each of the models containing the detailed progress for each of the MCMC runs. It also prints out a file (<resultsFilename>_model_cons.txt) that contains parameter values for the iterations with the lowest (negative) log likelihoods in numerical order.