
Protein crystallography
Research Interests
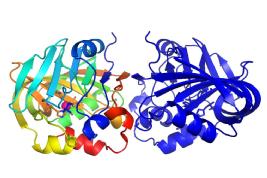
Oligopeptidases
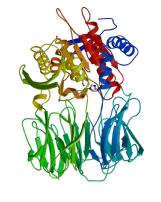
Prolyl oligopeptidase family enzymes regulate the activity of biologically active peptides and peptide hormones, and are implicated in diseases including amnesia, depression, diabetes and trypanosomiasis. Distinctively, these enzymes hydrolyze only relatively short peptide substrates, whilst large structured peptides and proteins are not usually cleaved. Prolyl oligopeptidase has a C-terminal alpha/beta-hydrolase catalytic domain that is similar to lipases and esterases. An N-terminal beta-propeller domain regulates access to the buried active site, explaining the observed oligopeptidase activity.
The catalytic and regulatory mechanisms have been investigated using a combination of X-ray crystallography, small-angle X-ray scattering (SAXS), site-directed mutagenesis, and enzyme kinetic measurements to provide better understanding of the structure-function properties of these physiologically and pharmaceutically important enzymes.
We are currently extending our structural studies on members of the peptidase family, which include enzymes of different specificities, such as dipeptidyl-peptidase IV, acylaminoacyl-peptidase, and oligopeptidase B; in order to facilitate a better understanding of the structure-function properties of these physiologically and pharmaceutically important enzymes.
Peroxidases and other haem proteins
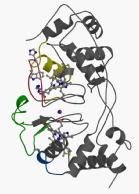
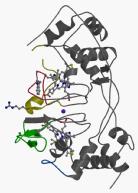
Bacterial cytochrome c peroxidases contain an electron transferring (E) haem domain and a peroxidatic (P) heme domain. All but one of these enzymes are isolated in an inactive oxidized state and require reduction of the E haem by a small redox donor protein in order to activate the P haem.
We have obtained the structures of the inactive oxidized and active mixed valence enzyme from both Pseudomonas aeruginosa and Paracoccus pantotrophus. The reduction of the E haem group might be coupled to proton uptake by its propionate group, in a process known as a redox-coupled Bohr effect, which in turn initiates a cascade of structural changes that prepares the enzyme for turnover. Chain flexibility in the former is strikingly distributed in certain loop regions, and these coincide with the regions of conformational change that occur in forming the active mixed valence enzyme. On the basis of these changes, we postulate a series of events that occur to link the trigger of the electron entering the E haem from either pseudoazurin or cytochrome c550 and the dissociation of a coordinating histidine at the P haem, which allows substrate access.
Our earlier investigations involved the Laue diffraction study on the structure of yeast cytochrome c peroxidise Compound I. We also obtained crystal structure of cytochrome cd1 nitrite reductase and we have trapped catalytic intermediates on the reaction pathway from the reduced to the oxidised state using flash freezing crystallographic studies combined with microspectrophotometric measurements. The changes observed both in haem ligation and conformational response of the enzyme upon ligand binding provided major insights into the mechanistic pathway.
The above research has general relevance to understanding of how electron transfer is coupled to structural changes and proton movements in proteins.
Enzymes involved in amino acid biosynthesis
Phosphoribosyl isomerase A (PriA)
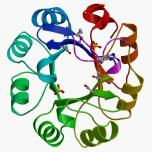
Recently we have solved two structures of phosphoribosyl isomerase A (PriA) from Streptomyces coelicolor, involved in both histidine and tryptophan biosynthesis. A closed conformer was obtained, which represents the first complete structure of PriA, revealing hitherto unnoticed molecular interactions and the occurrence of conformational changes. Inspection of these conformers, including ligand-docking simulations, allowed identification of residues involved in substrate recognition, chemical catalysis and conformational changes. These predictions were validated by mutagenesis and functional analysis. Arg19 and Ser81 were shown to play critical roles within the carboxyl and amino phosphate-binding sites, respectively; the catalytic residues Asp11 and Asp130 are responsible for both activities; and Thr166 and Asp171, which make an unusual contact, are likely to elicit the conformational changes needed for adopting the active site architectures. This represents the first report of the structure/function relationship of this (beta/alpha)8-isomerase.
The structural data will be complemented with kinetic/activity measurements. We anticipate that this will have an impact on the development of novel directed evolution approaches in vitro by means of mimicking natural evolution, as it has been established for the ((beta/alpha)8-barrel superfamily with enolase activity, and in drug design efforts targeting this enzyme present in pathogens, as previously suggested for other enzymes involved in these biosynthetic pathways in Mycobacterium species.
Acetolactate decarboxylase
Acetolactate decarboxylase (ALDC) is a bacterial enzyme of the butanediol fermentation pathway. ALDC has the unique ability to decarboxylate both
enantiomers of acetolactate to give a single enantiomer of the decarboxylation product, (R)-acetoin. The enzyme is widely used commercially by the brewing industry. It shortens the maturation step by preventing the formation of diacetyl, which gives an off taste to beer.
We have obtained the structure of Bacillus brevis ALDC using X-ray crystallography. Catalytic site of the enzyme contains a zinc ion coordinated by three conserved histidines and a glutamate. Currently we explore the chemical space of the active site using a series of chiral substrate analogues in order to to present a mechanism for the stereospecific decarboxylation of acetolactate. This will be further developed to novel applications to drive asymmetric chemical synthesis.