
Frenguelli Lab
Research Leader: Bruno FrenguelliLink opens in a new window
Novel Painkillers based on the Selective Activation of G protein Gα Subunits
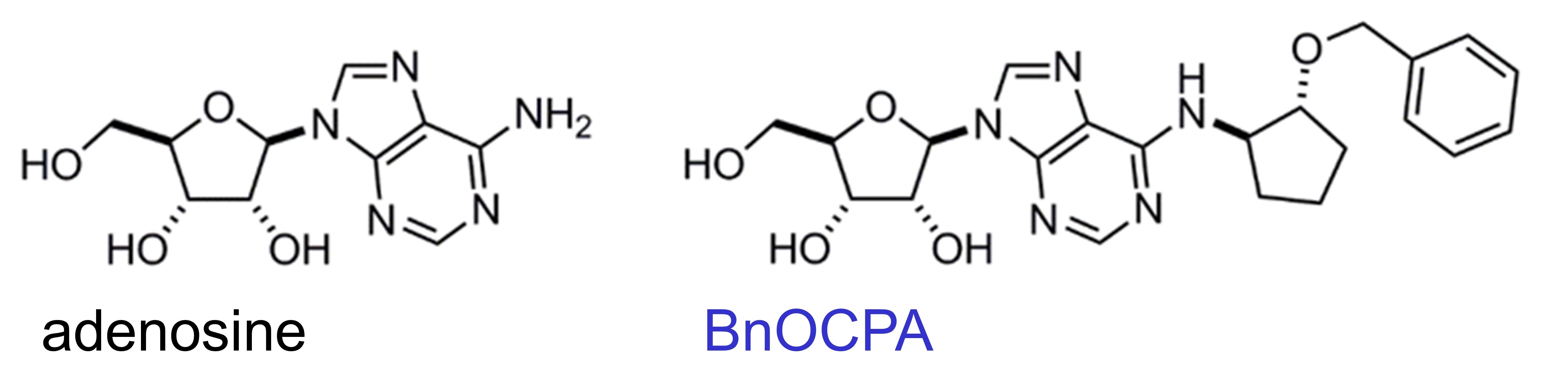
The purine nucleoside adenosine, a powerful endogenous regulator of neuronal activity, has long been known to have painkilling properties. However, adenosine also causes sedation, a drop in blood pressure, the slowing of the heart and respiratory depression. Much of the analgesia, sedation and cardiorespiratory effects of adenosine are caused by the activation of the adenosine A1 receptor (A1R), which has prevented the development of compounds that activate the A1R (agonists) as painkillers. However, we have recently described a molecule, BnOCPALink opens in a new window, synthesised by Dr Martin Lochner (U. Bern), that activates the A1R and induces analgesia but without the depressant effects on the heart, respiration and wakefulness.
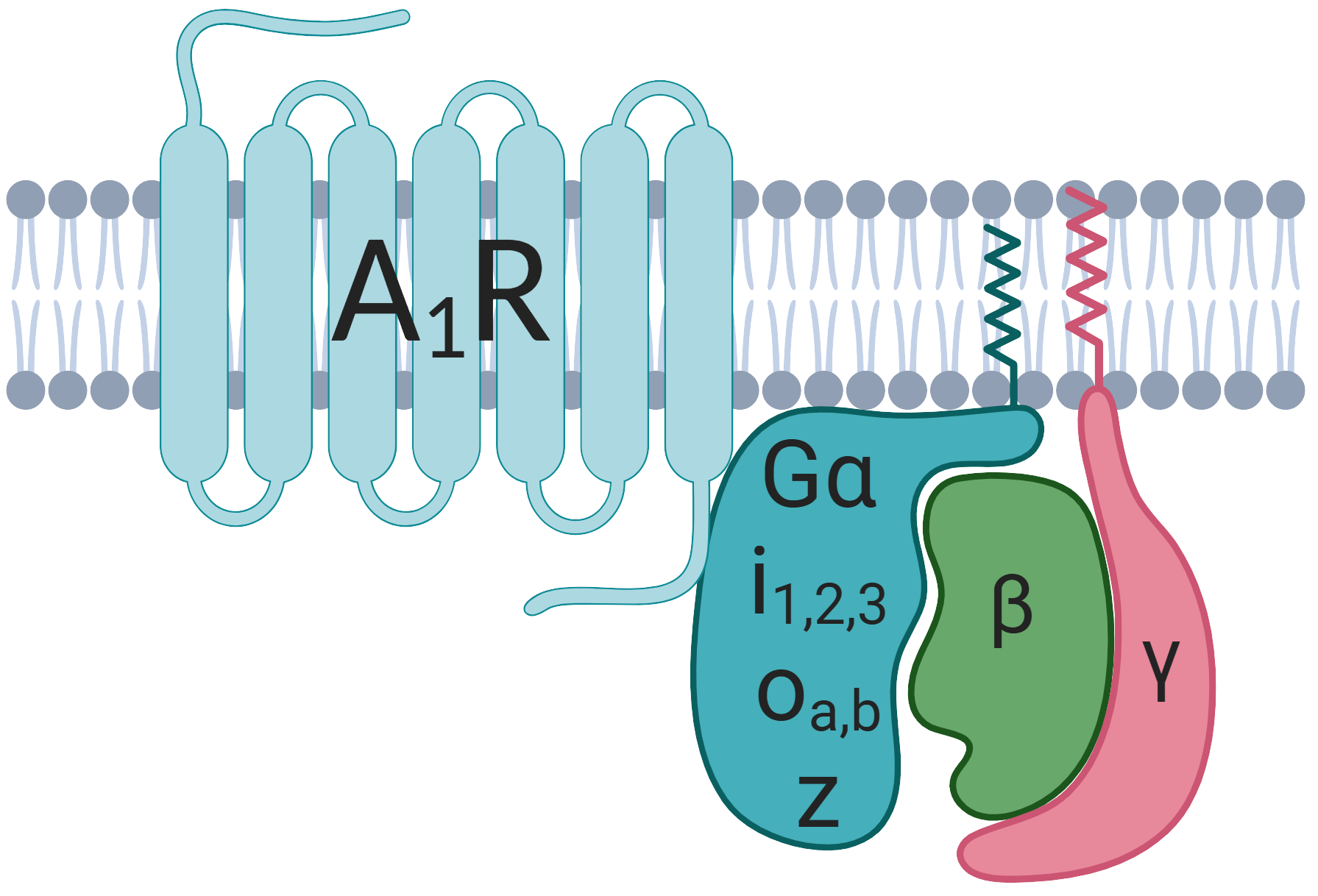
Our hypothesis for this peculiar and unprecedented observation is that BnOCPA only activates one of the six possible ways in which the A1R typically influences the behaviour of cells, tissues and organs such as the heart. Receptors such as the A1R are known as G protein-coupled receptors (GPCRs), because they need to couple to a Gα protein inside the cell to exert their actions. For the A1R there are six such Gα proteins - Gi1,2,3, Goa,b and Gz. From studies conducted by Prof Graham Ladds (U. Cambridge) BnOCPA only seems to activate the Gob subunit, so much so that BnOCPA seems to act like a blocking drug (antagonist) when the A1R is associated with the other subunits. This likely explains why BnOCPA does not affect the heart as the Gob subunit is not found at high levels in the heart, and why BnOCPA actually _blocks_ the effect of adenosine on the heart!

What might explain this unusual property of BnOCPA? From computer simulations performed by our collaborators Prof Chris Reynolds and Dr Giuseppe Deganutti (U. Coventry), of the binding of BnOCPA to the A1R it seems that BnOCPA can adopt one of four binding positions. Very peculiarly, two of these positions reflect conventional agonist binding, while the two others are more like the positions adopted by antagonists. Further computer simulations revealed that the coupling of the A1R to the Gα subunit was only productive for BnOCPA and Gob and not other Gα subunits. These properties of BnOCPA, which can be observed in intact physiological systems, such as those regulating pain perception and the cardiorespiratory systems, are very unusual and may be unique for currently available compounds that target the A1R and potentially receptors for other molecules. It does however set an important precedent that such selective targeting of Gα subunits is possible and could be deployed at receptors, like those for adenosine, that have attractive therapeutic potential as medicinal targets, but also have side effects that need to be avoided.
Potential Diagnostics and Treatments for Stroke
The mammalian brain is very sensitive to reductions in the supply of oxygen (hypoxia) and glucose (hypoglycemia). The large number of incidents during which one or other or both (ischemia) can occur (stroke, heart attack, head injury, near-drowning, carbon monoxide poisoning, hypoglycemia, epileptic seizures) indicates the scale of the problem. Intense efforts are being made to investigate the sequence of cellular and molecular events that are initiated by cerebral hypoxic/ischemic episodes with a view to providing better treatments.
Our laboratory is especially interested in the effects of hypoxia and ischemia on synaptic transmission in the mammalian hippocampus. The hippocampus is a region of the brain critically involved in certain forms of learning and memory, but, paradoxically, is one of the most vulnerable regions of the brain to hypoxia/ischemia. There are many human clinical cases studies in which cerebral ischemia is associated with profound anterograde amnesia and selective pathology of the hippocampus, in particular of CA1 pyramidal neurones.
To investigate the neuronal response to hypoxia/ischemia we use a combination of electrophysiological, pharmacological, biochemical, biosensor and imaging techniques.
Helping the brain help itself
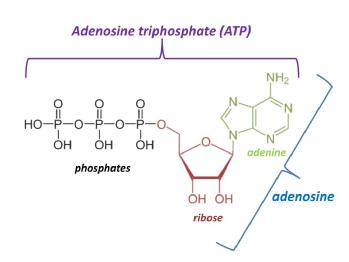
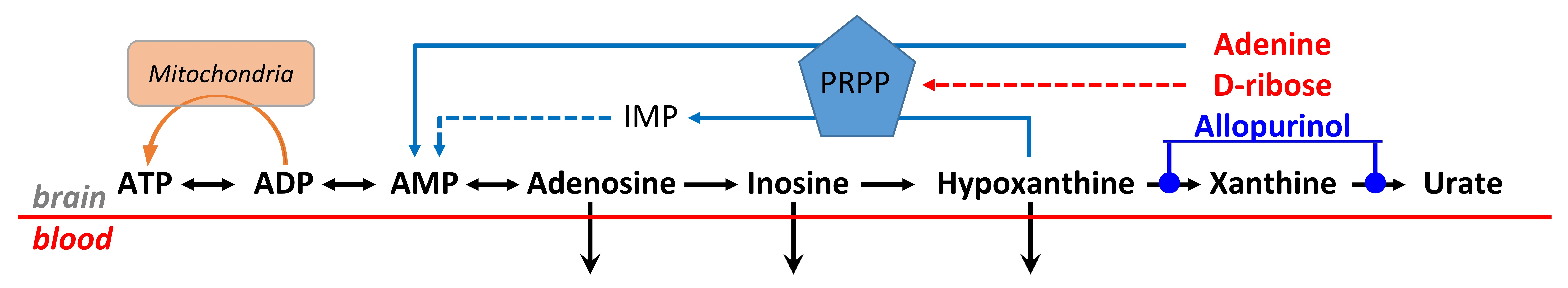
ATP (adenosine triphosphate) is the primary source of both cellular energy and the neuroactive metabolite adenosine. The breakdown of ATP during metabolic and traumatic stress results in the release of adenosine into the extracellular space. This has the beneficial effects of reducing neuronal activity (which would otherwise consume ATP) and dilates blood vessels in the brain, increasing the supply of nutrients. However, while this is beneficial to the brain in the short term, adenosine and its metabolites inosine and hypoxanthine can be lost from the brain into the general circulation. This is troublesome since the brain requires hypoxanthine to make ATP via the purine salvage pathway The loss of these metabolites to the blood stream likely explains the slow recovery of ATP levels in the injured brain. This would limit both the capacity of the brain to activate reparative processes and reduces the size of the adenosine reservoir, with implications for the development of post-injury seizure activity and the eventual recovery of the brain. However, their presence in blood could serve as a diagnostic biomarker for strokeLink opens in a new window, and we are examining this possibility using biosensors for adenosine and its metabolites in a models of stroke.
In order to test if the reduced ATP levels seen in brain tissue after a stroke could be increased, we used a very simple combination of test tube models of stroke and the building blocks of ATP. Using this approach we have shownLink opens in a new window that brain slices, which are known to have reduced ATP levels due to the ischemia and physical trauma associated with their preparation, can have their ATP levels increased to close to in vivo values. This is done by the simple addition of D-ribose and adenine (“RibAde”) to their incubation solution. D-ribose provides the sugar backbone of ATP, whilst adenine provides the nucleobase to which D-ribose is attached. Subsequent actions of a number of purine salvage enzymes convert the D-ribose and adenine into ATP. This process is more energy efficient compared to the de novo formation of ATP and is the dominant mechanism by which the brain makes the adenine nucleotide precursor to ATP, AMP.
The improved tissue content of ATP caused by RibAde has a number of functional consequences: the increased cellular ATP results in a larger reservoir of adenosine which can be released in response to physiological and pathologicalLink opens in a new window stimulation. Increased release of adenosine during high frequency (theta burst) stimulation raises the threshold for the induction of long-term potentiation (LTP), whilst during brief oxygen/glucose deprivation (OGD), increased adenosine is released which accelerates the inhibition of synaptic transmission and delays its recovery on reoxygenation. Furthermore, application of RibAde after OGD in cultured neurones reduced cell death, and reduces the intensity of seizure activityLink opens in a new window in hippocampal brain slices.
Extending these studies to an in vivo model of stroke, and in collaboration with colleagues at the university of Glasgow, we showed Link opens in a new windowthat the combination of ribose and adenine (RibAde) or ribose, adenine and allopurinol (RibAdeAll), which prevents the breakdown of hypoxanthine to xanthine and makes more hypoxanthine available for salvage, greatly reduced the brain damage associated with stroke and accelerated recovery in the week after the stroke. These data, plus the fact that D-ribose, adenine and allopurinol are well tolerated by humans, suggest that RibAde or RibAdeAll may be useful in patients having suffered from some form of brain injury, a hypothesis we are testing in models of ischemic stroke, the most common form of human stroke, with funding generously provided by the Stroke AssociationLink opens in a new window.
The Influence of Experience on the Brain
Long-Term Potentiation and learning and memory
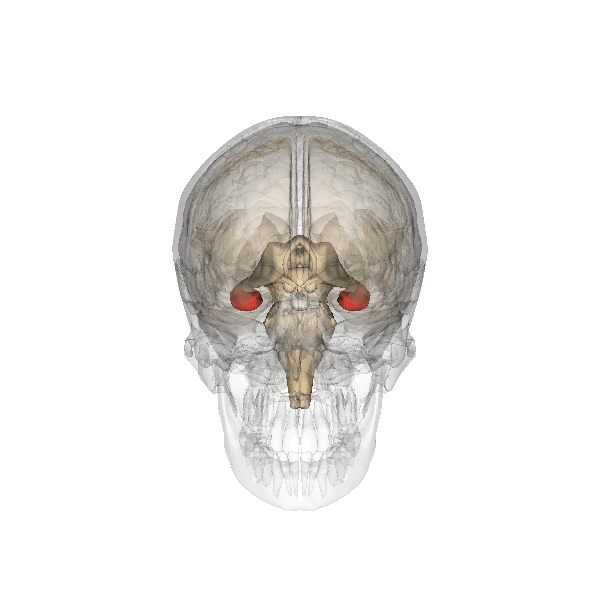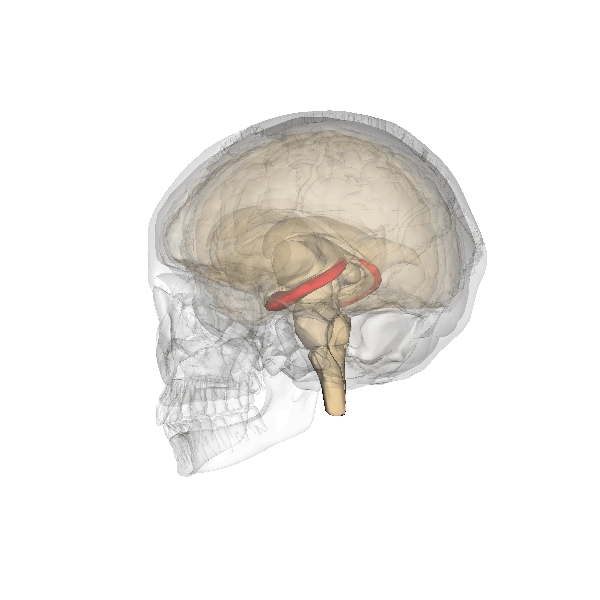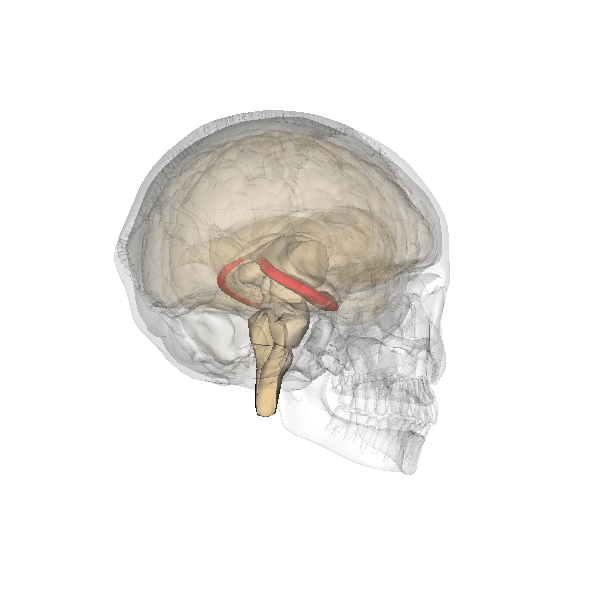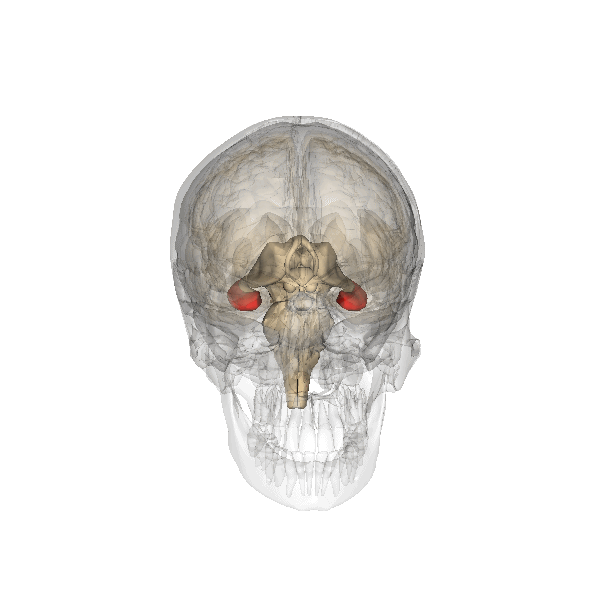
Synaptic plasticity is a process by which the strength of communication between neurones can be increased or decreased. Synaptic plasticity is believed to underlie the storage and recall of information, in other words, learning and memory. At present, the best cellular model for synaptic plasticity is long-term potentiation (LTP), a phenomenon that possesses many of the properties associated with learning and memory in mammals. A great deal of research has been undertaken on LTP in the hippocampus, a region of the brain (left; shown in red) especially involved in certain types of learning and memory. The hippocampus is affected in many types of insults to the brain and is one of the first brain regions to be damaged during the progress of Alzheimer’s disease and likely results in the forgetfulness of patients suffering from the early stages of Alzheimer’s.
Experience-dependent synaptic plasticity and the role of MSK1
Long-term changes in neuronal function underlie the behavioural and cognitive response to experience. Some of these experiences promote the rapid and persistent encoding of information and can have valuable repercussions – for example the association of illness or death (of others!) with poisonous plants or animals, in order to avoid them in future. On a more mundane level, such associations allow us to remember where we left our house key, mobile phone or that we have an appointment to keep or a lecture to go to. However, sensory experiences can occur over prolonged periods – consider learning to read or write or play a musical instrument. In both these cases of rapid and protracted acquisition, long-term storage or adaptation requires changes at the genomic level to provide the proteins necessary to hard wire these experiences. As such, the genomic changes, and the mechanisms by which they are induced have been the subject of considerable investigation.
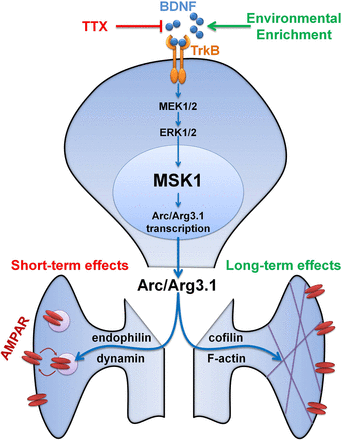
We have been examining the contribution of a protein kinase – MSK1 – to experience and protracted activity-dependent synaptic plasticity. MSK1 is interesting as it is activated by BDNF, a key growth factor in the brain implicated in a range of processes from brain development to learning and memory. Moreover, MSK1 exerts is actions via influencing gene transcription through the phosphorylation of the transcription factor CREB and the chromatin-associated histone H3. Accordingly, MSK1 is well-placed to transduce the effects of BDNF into long-lasting changes in neuronal structure and function.
We have shownLink opens in a new window that MSK1 is necessary for the regulation of synaptic strength in response to prolonged inactivation of synapses caused by blocking voltage-gated sodium channels with TTX. Moreover, MSK1 is also necessary for the enhancement of synaptic transmission associated with environmental enrichment, in which rodents are raised in an environment providing them with additional social and sensory stimulation through the provision of larger numbers of cage-mates, ladders, rope swings, tubes and exercise wheels. Subsequent studiesLink opens in a new window have shown that MSK1 is necessary for the extension of the dynamic range of synapses in that they can both strengthen more (greater LTP) and weaken more (greater long-term depression; LTD) and that MSK1 is required for more complex and cognitively demanding learning (reversal learning) and the persistence of memory. Indeed, these actions of MSK1 persist through the life-span into old age. Link opens in a new windowThese studies Link opens in a new windowalso revealed the unexpected and MSK1-dependedent downregulation of genes associated with learning and memory. One potential explanationLink opens in a new window is that since it is not practical to continue expressing genes, the downregulation "resets" gene expression levels to a lower baseline such that when a new experience comes along, the expression of new genes is more readily apparent to deliver a greater impact. MSK1 thus seems to serve a very important role in the regulation, or homeostasis, of the activity of the nervous system.
Prompted by our studies of the influence of the environment on the brain and behaviour, and the rich, if sometimes depressing, human literature on this topic, I decided to launch a new journal - Brain and EnvironmentLink opens in a new window - specifically aimed at considering the importance of gene x environment interactions and the impact of nurture on nature. You can read a Q&ALink opens in a new window with me and Deputy Editor, Dr Margaret Sheridan, and the Editorial Link opens in a new windowwe wrote on our aspirations for the new journal.
Stuff I've been getting up toLink opens in a new window
Join the Lab!Link opens in a new window
Lab membersLink opens in a new window
CollaboratorsLink opens in a new window
FundingLink opens in a new window
PublicationsLink opens in a new window